Zhang, K., Zemke, N.R., Armand, E.J. et al. A fast, scalable and versatile tool for analysis of single-cell omics data. Nat Methods 21, 217–227 (2024).
论文地址:https://doi.org/10.1038/s41592-023-02139-9
代码地址:https://github.com/kaizhang/SnapATAC2/
摘要
单细胞组学技术在复杂组织中基因调控研究领域带来了革命性进展。分析这些数据集的一大计算挑战在于,将大规模、高维数据投影到低维空间的同时,保留细胞之间的相对关系。这种低维嵌入对于分解细胞异质性和重建细胞类型特异的基因调控程序至关重要。然而,传统的降维技术在计算效率和全面处理不同分子模式下的细胞多样性方面面临挑战。为此,我们引入了一种非线性降维算法,并将其整合到Python包SnapATAC2中。该算法不仅能够更精确地捕捉单细胞组学数据的异质性,还能确保运行效率和内存使用的优化,其性能随细胞数量线性扩展。我们的算法在多种单细胞组学数据集上表现出卓越的性能、可扩展性和多功能性,包括单细胞转座酶可及染色质测序(scATAC-seq)、单细胞RNA测序(scRNA-seq)、单细胞Hi-C以及单细胞多组学数据集。这充分表明了其在推动单细胞分析领域发展的实用价值。
引言
单细胞组学技术的快速发展,使得对基因组中编码的基因调控程序的分析达到了前所未有的分辨率和规模。单细胞基因组、转录组、开放染色质图谱、组蛋白修饰、转录因子结合位点、DNA甲基化和染色质结构等的分析,为理解细胞身份和调控机制提供了宝贵的见解。然而,单细胞组学数据的极大规模和复杂性常常带来显著的计算挑战,迫切需要开发高效、可扩展且鲁棒性强的数据分析方法。
分析单细胞组学数据的一个关键步骤是将高维数据投影到低维空间,同时保留细胞之间的相对关系,这一过程被称为降维。降维对于后续分析(如聚类、批次效应校正、数据整合和可视化)的成功至关重要。有效的降维技术是可视化不同细胞群体、识别稀有细胞类型以及描绘细胞类型特异性转录调控程序的基础。目前,单细胞组学降维算法主要分为线性和非线性两类。线性降维算法如主成分分析(PCA),被SCANPY和Seurat用于单细胞RNA测序(scRNA-seq)数据分析;潜在语义索引(LSI)被ArchR和Signac用于单细胞开放染色质测序(scATAC-seq)数据分析。这些算法因其计算效率和可扩展性而广受欢迎,但在处理具有复杂非线性结构的数据集(如单细胞Hi-C和单细胞多模态组学数据集)时并不理想。
非线性降维方法则更擅长捕捉复杂且常常是非线性的细胞关系。例如,潜在狄利克雷分布(LDA)被用于scATAC-seq和scHi-C数据;基于拉普拉斯的算法被用于scRNA-seq和scATAC-seq数据;各种用于scRNA-seq、scATAC-seq和scHi-C数据的神经网络模型。这些非线性降维方法已成为单细胞数据可视化的标准方法,例如t分布邻域嵌入(t-SNE)和统一流形近似与投影(UMAP)尽管最近对其可靠性和有效性提出了一些质疑。尽管非线性方法在处理复杂结构和低维流形投影方面表现出色,但通常计算效率较低且可扩展性有限。例如,LDA依赖于马尔科夫链蒙特卡罗算法进行模型训练,收敛速度慢,计算成本高且难以并行化,使其难以应用于大型数据集。基于拉普拉斯的技术(如我们之前的SnapATAC工作)需要计算所有细胞对之间的相似性矩阵,导致内存使用随细胞数量呈二次增长。深度神经网络模型虽然性能强大,但训练成本高,通常需要图形处理单元(GPU)等专用硬件支持。
在本研究中,我们提出了一种非线性降维算法,能够在解析复杂组织的细胞组成时兼顾计算效率和准确性。我们的关键创新在于使用矩阵无关的谱嵌入算法,将单细胞组学数据投影到保留数据内在几何特性的低维空间。不同于传统的谱嵌入方法(需要构建图拉普拉斯矩阵,存储需求随细胞数量呈二次增长),我们的算法通过使用Lanczos算法隐式操作拉普拉斯矩阵,避免了这一计算昂贵的步骤。这一策略显著降低了时间和空间复杂度,使其随单细胞数据的细胞数量线性扩展。
为了评估算法的准确性和实用性,我们在包含不同实验方案、物种和组织类型的多种数据集上进行了广泛的基准测试。结果表明,我们的矩阵无关谱嵌入算法在速度、可扩展性和解析细胞异质性方面均优于现有方法。此外,我们展示了该算法可扩展到多种单细胞组学数据的分子模式,利用来自不同单细胞组学数据类型的互补信息揭示细胞异质性。
我们将这些算法改进实现为一个Python包SnapATAC2。这是对原始SnapATAC的重大升级,提供了显著的改进,包括更快的速度、更低的内存使用、更可靠的性能以及面向多种单细胞组学数据的综合分析框架。SnapATAC2可从 https://github.com/kaizhang/SnapATAC2/ 免费获取。
SnapATAC2 工作流程概览
SnapATAC2 是一种全面且高性能的单细胞组学数据分析解决方案。与原版 SnapATAC 相似,SnapATAC2 提供了丰富的功能,覆盖 scATAC-seq 数据分析流程的多个阶段。此外,SnapATAC2 的设计具有高度灵活性,可适用于多种单细胞组学数据类型。例如,其降维子程序不仅可用于 scATAC-seq 数据,还能适配 scRNA-seq、单细胞 DNA 甲基化和 scHi-C 数据,体现了其强大的适应性。
为了提升性能和可扩展性,SnapATAC2 使用了 Rust 编程语言来执行计算密集型子程序,并提供了 Python 接口以实现便捷的安装和友好的用户体验。这种组合使得 SnapATAC2 能高效处理大规模单细胞组学数据,同时适用于不同研究背景的用户。此外,为了进一步提高在处理大规模单细胞数据时的可扩展性,SnapATAC2 尽可能采用磁盘数据结构和外存算法,使其在分析大型数据集时不至于过度占用系统资源。
SnapATAC2 具有模块化和可定制性,用户可以根据特定需求调整分析流程,同时还能与 scverse 生态系统中的其他软件包(如 SCANPY 和 scvi-tools)无缝集成,从而增强其适用性和功能性。
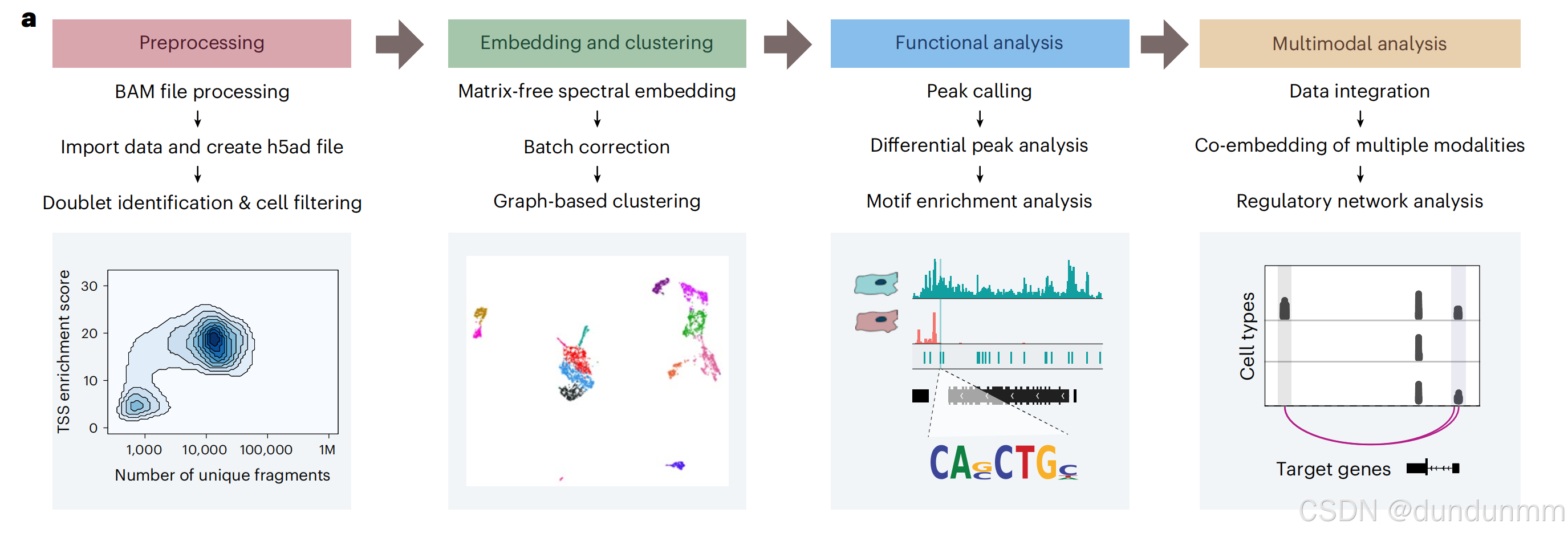
SnapATAC2 包括四个主要模块:预处理、嵌入/聚类、功能富集分析以及多模态组学分析(如图 1a 所示)。
- 预处理模块 负责处理原始 BAM 文件、评估数据质量、生成计数矩阵以及识别双重细胞,为后续分析奠定了坚实基础。
- SnapATAC2 的核心是其 嵌入/聚类模块,其中引入了一种新的降维算法,用于识别独特的细胞簇并揭示生物学模式。
- 功能富集模块 提供了详细的数据解读,包括差异可及性分析和基序分析。
- 多模态组学分析模块 则允许研究人员整合多种生物数据,分析复杂且多面的生物学数据集,并构建基因调控网络以加深对生物学机制的理解。
讨论
在本研究中,我们介绍了用于分析多种单细胞组学数据的工具 SnapATAC2。相比现有的降维方法,SnapATAC2 在准确性、抗噪性和可扩展性方面表现更为出色,为研究者提供了强大的工具来通过单细胞基因组学、转录组学和表观基因组学分析探究基因调控程序。
SnapATAC2 的一个显著优势是其与单细胞分析生态系统中广泛使用的软件工具的无缝兼容性。通过采用 AnnData 数据格式,SnapATAC2 能轻松与 SCANPY、scvi-tools 和 SCENIC+ 等成熟软件包集成。这一功能对于需要进行专业分析(如数据插补或轨迹推断)的研究者尤为有利,从而进一步增强了 SnapATAC2 的核心功能。
SnapATAC2 的关键创新在于其 基于矩阵自由光谱嵌入算法的降维方法。尽管已有许多算法被提出用于加速光谱嵌入,但 SnapATAC2 的算法独树一帜,因为它不依赖于子采样或近似计算,而是提供精确解。该算法不仅在细胞聚类和异质性识别方面优于现有方法,还保持了计算效率,非常适合于大规模单细胞组学数据分析。此外,我们通过将该矩阵自由光谱嵌入算法应用于多种单细胞数据类型(包括 scATAC-seq、scRNA-seq、单细胞 DNA 甲基化、scHi-C 和单细胞多组学数据),验证了其通用性。
算法局限性
目前,矩阵自由光谱嵌入算法仅基于余弦相似度计算。然而,对于某些数据类型,研究者可能更倾向于使用其他度量方法来量化细胞间的相似性。例如,我们的研究发现,对于在转录组和表位索引实验中使用的蛋白表达数据,欧几里得距离能够提供更准确的结果。未来的开发可以扩展矩阵自由算法以适应其他相似性度量方法。一种潜在的解决方案是利用一小组标记点将数据转化为稀疏特征向量,然后应用可扩展的矩阵自由光谱嵌入算法。
综上,SnapATAC2 在单细胞数据分析领域代表了一项重要进展,为研究表观基因组学提供了一种易于使用、可扩展且高性能的解决方案。随着持续的开发与优化,SnapATAC2 有潜力成为单细胞多组学数据分析的通用工具,最终推动新的生物学发现的实现。
模型的创新点在于在降维计算阶段降低了计算复杂度且具有可扩展性,后续将关注具体的计算复杂度