PLIP: 如何分析蛋白质和配体相互作用?
蛋白质和配体之间的相互作用就像一把钥匙开一把锁,只有完美契合才能发挥最佳效果。想要设计出高效的药物,深入了解这些相互作用至关重要。
今天,我们将以 PD-L1 和几种小分子配体的共晶结构为例,带你一步步利用 PLIP 和 PLIF 等工具,了解蛋白质和配体相互作用。
读完本文,你将学习到:
- 了解 PLIP 和 PLIF 的基本概念及应用。
- 掌握利用 RDKit 分析 PLIF 数据的方法。
- 学习如何识别关键氨基酸残基以及它们参与的相互作用类型。
引言
我们希望能够以某种更直观的方式利用蛋白和配体结合的信息。在一番搜索之后,找到了一个可以识别相互作用分析方法:Protein-Ligand Interaction Profiler (PLIP)。
PLIP 的文献:Adasme, Melissa F., et al. “PLIP 2021: expanding the scope of the protein–ligand interaction profiler to DNA and RNA.” Nucleic Acids Res., vol. 49, no. W1, 2 July 2021, pp. W530-W534, doi:10.1093/nar/gkab294.
PLIP 看起来能够很好地分析蛋白配体相互作用。
PLIP?
首先,我们来看一下 PLIP 的网页界面:
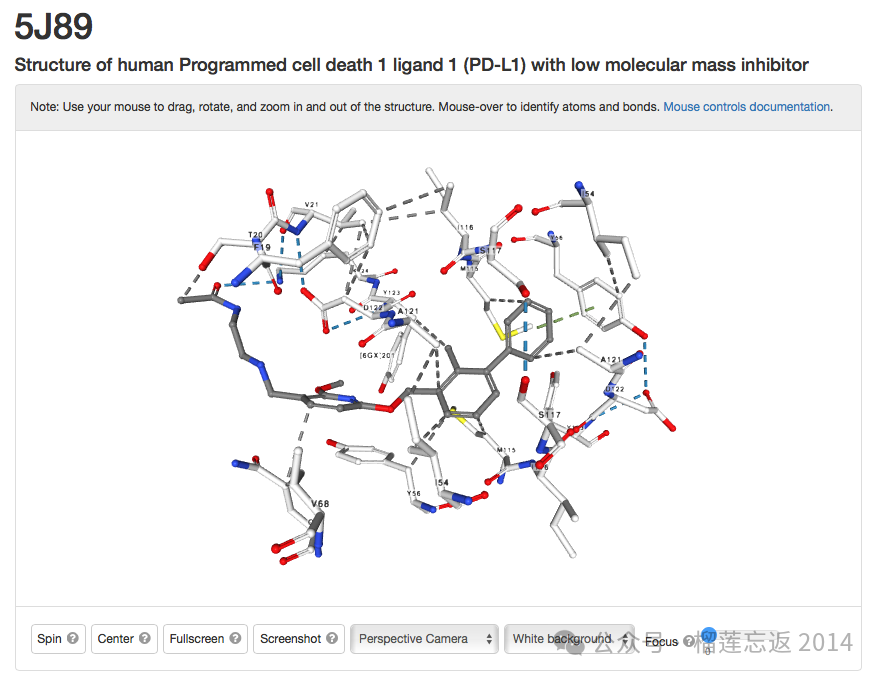
尝试着将复合体(PDB ID: 5J89)提交到 PLIP 中进行分析,结果如下:
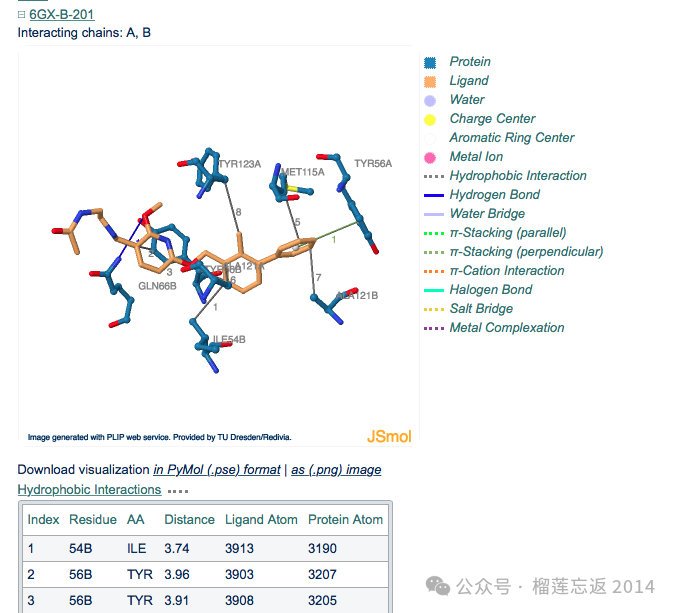
PLIP 看起来能够很好地将相互作用分类并列出来。分析结果可以下载下来,并用 PyMol 进行查看。真心不错!
识别交互作用的类型
根据 Supplementary Information,默认情况下,PLIP 可以识别以下非共价相互作用:
| 相互作用 | 英文 | 基准 | 变量 |
|---|---|---|---|
| 疏水相互作用 | Hydrophobic interactions | 距离 (≤ 4.0Å) | HYDROPH_DIST_MAX |
| 氢键 | Hydrogen Bonds | 距离 (≤ 4.1Å) 角度 (≥ 100°) | HBOND_DIST_MAX HBOND_DON_ANGLE_MIN |
| π-π 堆积 | Aromatic Stacking | 距离 (≤ 7.5Å) 角度 (T-stacking 90°±30°, P-stacking 180°±30°) 环中心距离 (≤ 2.0Å) | PISTACK_DIST_MAX PISTACK_ANG_DEV PISTACK_OFFSET_MAX |
| π-阳离子相互作用 | Pi-Cation interactions | 距离 (≤ 6.0Å) (三级胺需要考虑角度) | PICATION_DIST_MAX |
| 盐桥 | Salt Bridges | 电荷中心距离 (≤ 5.5Å) | SALTBRIDGE_DIST_MAX |
| 水介导的氢键 | Water-brodged hydrogen bonds | 水分子位置 (2.5Å~4.0Å) 角度 (75°<ω<140°, 100°<θ) ) | WATER_BRIDGE_MINDIST, WATER_BRIDGE_MAXDIST WATER_BRIDGE_OMEGA_MIN, WATER_BRIDGE_OMEGA_MAX WATER_BRIDGE_THETA_MIN |
| 卤键 | Halogen bonds | 距离 (≤ 4.0Å) 角度 (Donor 165°±30°, Acceptor 120°±30°) | HALOGEN_DIST_MAX, HALOGEN_DON_ANGLE, HALOGEN_ACC_ANGLE, HALOGEN_ANG_DEV |
本地安装 PLIP
安装环境要求
PLIP 可以从命令行使用,赶紧尝试在我的电脑上安装它。
根据 Github 上的说明[1],PLIP 使用 OpenBabel 来识别原子属性,因此需要安装 OpenBabel(>= 2.3.2)。此外,PLIP 还依赖于以下可选软件包:
- PyMOL(>=1.7.x 且支持 Python bindings)
- Imagemagick(>=1.7.x)
- swig
在 Python 2.7.x 环境下,可以使用以下命令安装 PLIP:
pip install plip
但是,在尝试使用 pip3 进行安装时,遇到了 OpenBabel wheel 文件相关的错误。
安装过程中遇到的问题及解决方法
在运行 PLIP 时遇到了以下错误:
File "/usr/local/lib/python3.7/site-packages/plip/modules/supplemental.py", line 388, in read_pdb
resource.setrlimit(resource.RLIMIT_STACK, (min(2 ** 28, maxsize), maxsize))
ValueError: current limit exceeds maximum limit
不太明白错误的原因,查看相应的代码可以发现:
def read_pdb(pdbfname, as_string=False):
"""Reads a given PDB file and returns a Pybel Molecule."""
pybel.ob.obErrorLog.StopLogging()
if os.name != 'nt':
maxsize = resource.getrlimit(resource.RLIMIT_STACK)[-1]
resource.setrlimit(resource.RLIMIT_STACK, (min(2 ** 28, maxsize), maxsize))
sys.setrecursionlimit(10 ** 5)
return readmol(pdbfname, as_string=as_string)
这段代码是一个读取 PDB 文件的函数。根据 Python 资源库 resource[2] 的说明,报错的代码似乎是在限制程序使用的系统资源。如果限制的是读取 PDB 文件的资源,那么应该可以注释掉这段代码。
修改后的代码如下:
def read_pdb(pdbfname, as_string=False):
"""Reads a given PDB file and returns a Pybel Molecule."""
pybel.ob.obErrorLog.StopLogging()
sys.setrecursionlimit(10 ** 5)
return readmol(pdbfname, as_string=as_string)
保存文件后再次运行,程序不再报错,并成功输出了 XML 文件。虽然不确定这样做是否妥当,但至少程序可以正常运行了。
另外还有一种很好的办法,那就是使用:docker 。它非常简单,一行命令就能搞定。这里就不再赘述。
分析共晶体结构数据
这次我想要分析的共晶体结构是 PD-L1 和小分子的共晶体结构。整理小分子的信息如下:
| PDB entry | Resolution (Å) | Chain | Positions | 配体 | 配体 ID | 文献 |
|---|---|---|---|---|---|---|
| 5J89 | 2.20 | A/B/C/D | 2-134 | BMS-202 | 6GX | Oncotarget 2016(7)30323 |
| 5J8O | 2.30 | A/B | 18-134 | BMS-8 | 6GZ | Oncotarget 2016(7)30323 |
| 5N2D | 2.35 | A/B/C/D | 2-134 | BMS-37 | 8J8 | J. Med. Chem. 2017(60)5857 |
| 5N2F | 1.70 | A/B | 18-134 | BMS-200 | 8HW | J. Med. Chem. 2017(60)5857 |
| 5NIU | 2.01 | A/B/C/D | 18-134 | BMS-1001 | 8YZ | Oncotarget 2017(8)72167 |
| 5NIX | 2.20 | A/B/C/D | 18-134 | BMS-1166 | 8YQ | Oncotarget 2017(8)72167 |
运行分析
命令行使用方法
PLIP 可以通过 PDB ID 从 PDB 服务器上获取数据并进行分析,因此在终端中运行以下命令:
alias plip='python ~/pliptool/plip/plipcmd.py'
plip -i 5J89 -x
-i PDBID 用于指定要分析的结构的 ID,-x 用于将结果输出为 XML 报告文件。
RDKit 与 PLIF 计算
PLIF 是什么?
PLIF(Protein-Ligand Interaction Fingerprints)是一种使用指纹描述配体-受体相互作用类型和强度的统计方法,用于分析多个结合状态。
它可以分析对接结果或多个复合物结构中包含的相互作用,以:
- 检测与活性/非活性相关的相互作用
- 提取对活性/非活性进行分类的相互作用组合规则
- 检测符合规则的活性 pose 共有的药效团
其他类似方法
除了 PLIF 之外,还有其他类似的方法,例如:
- SIFt (JMC2004(47)337[3])
- aPLIF
- aPLIED
- Pharm-IF (JCIM2010(50)170[4])
分析之提取信息
import xml.etree.ElementTree as ET
def generate_plif_lists(report_file, residue_list, lig_ident):
plif_list_all = []
tree = ET.parse(report_file)
root = tree.getroot()
for binding_site in root.findall('bindingsite'):
nest = binding_site.find('identifiers')
lig_code = nest.find('hetid')
if str(lig_code.text) == str(lig_ident):
nest_residue = binding_site.find('bs_residues')
residue_list_tree = nest_residue.findall('bs_residue')
for residue in residue_list_tree:
res_id = residue.text
dict_res_temp = residue.attrib
if res_id not in residue_list:
residue_list.append(res_id)
if dict_res_temp['contact'] == 'True':
if res_id not in plif_list_all:
plif_list_all.append(res_id)
return plif_list_all, residue_list
bs_res_list = []
P_L_dict = {"5J89":"6GX","5J8O":"6GZ","5N2D":"8J8",
"5N2F":"8HW","5NIU":"8YZ","5NIX":"8YQ"}
contact_res_dict = {}
for pdb_id, lig_id in P_L_dict.items():
pl = "conres_" + pdb_id
xml_path = "PLIP_results/" + pdb_id + "/report.xml"
pl, bs_res_list = generate_plif_lists(xml_path, bs_res_list, lig_id)
contact_res_dict[pdb_id] = pl
print(contact_res_dict.keys())
print(contact_res_dict['5J89'])
print(len(bs_res_list))
结果分析:结合位点氨基酸残基
- 代码首先定义了一个函数
generate_plif_lists,用于从 PLIP 的分析结果(XML 文件)中提取信息。 - 然后创建了一个空列表
bs_res_list,用于存储结合位点的所有氨基酸残基,以及一个字典P_L_dict,用于存储 PDB ID 和配体 ID 的对应关系。 - 接下来,代码遍历
P_L_dict中的所有键值对,并调用generate_plif_lists函数,将结果存储在contact_res_dict字典中。 contact_res_dict字典的键是 PDB ID,值是与配体接触的残基列表。- 最后,代码打印了
contact_res_dict字典的键、contact_res_dict['5J89']的值,以及bs_res_list的长度。
结果显示,bs_res_list的长度为 136,表明结合位点包含 136 个氨基酸残基。
共晶体结构中的配体似乎以 PD-L1 二聚体为中心对称排列。如果确实是这种情况,那么氨基酸序列的位置就比区分 CHAIN 更重要。因此,我们尝试在不区分 CHAIN 信息的情况下重新进行分析。
实际上,这只需要从提取信息中提取的残基信息中删除最后一个字母即可。
bs_res_list_num = []
for i in bs_res_list:
# 去除CHAIN信息
j = i[:-1]
bs_res_list_num.append(int(j))
# 去除重复信息并排序
bs_res_list_num_unique = sorted(set(bs_res_list_num))
print(len(bs_res_list_num_unique))
print(bs_res_list_num_unique)
接下来,我们从相互作用的残基信息中删除 CHAIN 信息,并去除重复信息。
contact_res_dict_num_unique = {}
for k, v in contact_res_dict.items():
tmp_list = []
for i in v:
# 从相互作用残基中去除CHAIN信息
j = i[:-1]
tmp_list.append(int(j))
tmp_unique = sorted(set(tmp_list))
contact_res_dict_num_unique[k] = tmp_unique
获取比特列表
直接使用 RDKit 的 BitVect 并将其转换回 numpy 数组会导致残基编号信息的丢失。为了解决这个问题,创建一个新的函数来生成比特列表。
def PLIF_list_generator(bs_residues, contact_residues):
tmp = []
for i in bs_residues:
if i in contact_residues:
tmp.append(1)
else:
tmp.append(0)
return tmp
然后,我们创建一个 DataFrame,其中行表示 PDB ID,列表示残基编号。
FP_df_num = pd.DataFrame(index=PDB_id_list, columns=bs_res_list_num_unique)
for k, v in contact_res_dict_num_unique.items():
FP_df_num.loc[k] = PLIF_list_generator(bs_res_list_num_unique, v)
结果如下:
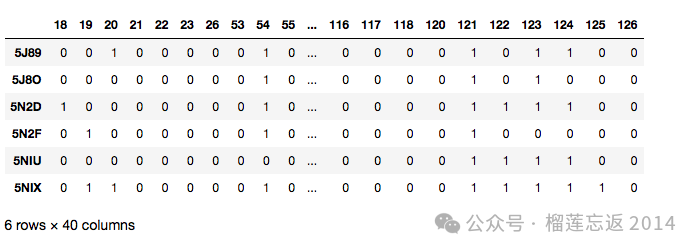
为了方便观察哪些残基在多个共晶体结构中都参与了相互作用,我们计算每个残基的比特和。
FP_df_num.loc['bit_sum']=FP_df_num.sum()
最后,使用热图可视化结果:
%matplotlib inline
sns.heatmap(FP_df_num)
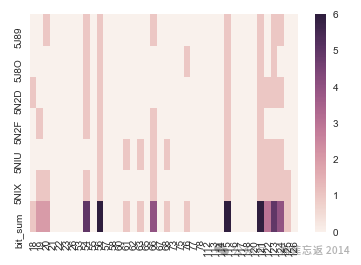
寻找重要残基
根据之前的分析,6 个共晶体结构可以分为 3 组。因此,bit_sum大于等于5的残基意味着它至少在 3 组中的每一组中都出现过一次。
Frequent_residues = list(FP_df_num.T.query('bit_sum >= 5').index)
print(Frequent_residues)
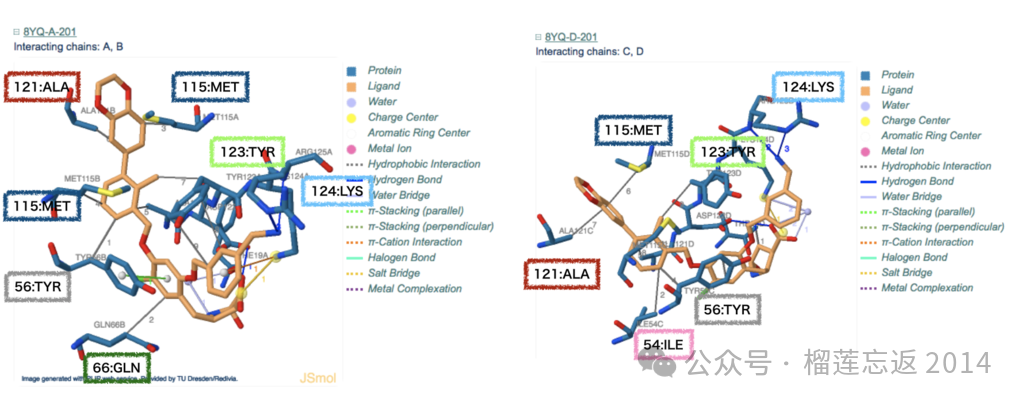
这些残基具体涉及哪些相互作用呢?我们以包含所有这些残基的PDB ID: 5NIX (配体: 8YQ)为例,使用 PLIP 网站进行分析。
5NIX是一个「PD-L1 4: Ligand 2」的复合体,包含两个配体。分析结果如下表所示:
| 残基编号 | 氨基酸 | 距离 | 相互作用 | 残基编号 | 氨基酸 | 距离 | 相互作用 | ||
|---|---|---|---|---|---|---|---|---|---|
| 8YQ | |||||||||
| (chain A/B) | 8YQ | ||||||||
| (chain C/D) | 54C | ILE | 3.89 | 疏水相互作用 | |||||
| 56B | TYR | 3.82 | 疏水相互作用 | 56C | TYR | 3.42/3.62 | 疏水相互作用 | ||
| 115A/B | MET | 3.70/3.80 | 疏水相互作用 | 115C/D | MET | 3.85/3.82 | 疏水相互作用 | ||
| 121A/B | ALA | 3.69/3.75 | 疏水相互作用 | 121C/D | ALA | 3.96/3.57 | 疏水相互作用 | ||
| 123A | TYR | 3.80/3.52/3.85 | 疏水相互作用 | 123D | TYR | 3.72 | 疏水相互作用 |
分析结果显示,除了预期的与配体芳香环的 π-π 相互作用外,还观察到多个与甲硫氨酸和丙氨酸的相互作用。
接下来,我们将bit_sum的门槛降低到 4,看看会有什么新的发现。
Frequent_residues4 = list(FP_df_num.T.query('bit_sum >= 4').index)
print(Frequent_residues4)
结果中新增了残基66和124,对应如下表所示:
| 残基编号 | 氨基酸 | 距离 | 相互作用 | 残基编号 | 氨基酸 | 距离 | 相互作用 | ||
|---|---|---|---|---|---|---|---|---|---|
| 8YQ | |||||||||
| (chain A/B) | 66B | GLN | 3.72 | 疏水相互作用 | 8YQ | ||||
| (chain C/D) | |||||||||
| 124A | LYS | 5.03/3.18 | π-阳离子/盐桥 | 124D | Lys | 3.70 | 水桥/盐桥 |
除了疏水相互作用外,新增了亲水性残基 Lys,使相互作用类型更加多样化。
以下是 PLIP 分析结果的截图,展示了这些相互作用的具体情况:
总结
本文介绍了蛋白质-配体相互作用指纹(PLIF)方法,该方法结合了蛋白质-配体相互作用分析软件(PLIP)和化学信息学工具包 RDKit,用于分析蛋白质-配体相互作用。
首先,利用 PLIP 软件分析了 6 个 PD-L1 共晶体结构,获得了配体与蛋白质残基之间的相互作用信息。然后,根据 OPIG 博客介绍的方法,利用 RDKit 将 PLIP 的分析结果转换为 PLIF。
为了便于比较不同共晶体结构之间的相互作用模式,将 PLIF 表示为位向量,并使用热图可视化。结果表明,根据位向量的分布,6 个共晶体结构可以分为 3 组,这与配体的分子量和蛋白质链的数量有关。
为了进一步分析,去除了蛋白质链信息,仅根据残基编号重新生成了 PLIF。结果表明,残基 54、56、115、121、123 在所有 6 个共晶体结构中都参与了相互作用,而残基 66 和 124 在至少 4 个结构中参与了相互作用。
通过分析 PLIP 的交互图,发现这些残基主要参与了疏水相互作用、π-阳离子相互作用、盐桥和水桥等相互作用。
PLIF 方法可以用于分析多个共晶体结构,识别关键的蛋白质-配体相互作用残基,并为药物设计提供指导。未来可以进一步探索 PLIF 在以下方面的应用:
- 结合虚拟筛选结果,优化化合物库的设计和筛选策略。
- 建立定量构效关系模型,预测化合物的活性。
- 分析蛋白质-蛋白质相互作用,研究蛋白质的功能和调控机制。