Large Language Models for Bioinformatics

目录
0. 摘要
随着大型语言模型(LLM)技术的快速发展以及生物信息学专用语言模型(BioLMs)的兴起,对当前领域的综合分析、计算特性及多样化应用的需求日益增长。
本综述旨在通过对 BioLMs 的演化、分类和独特特征的深入审查,解决这一需求,同时详细探讨其训练方法、数据集和评估框架。
我们研究了 BioLMs 在疾病诊断、药物发现和疫苗开发等关键领域的广泛应用,突出了其在生物信息学中的影响和变革潜力。
此外,我们识别了 BioLMs 固有的主要挑战和局限性,包括数据隐私与安全问题、可解释性问题、训练数据和模型输出中的偏差以及领域适应性复杂性。
最后,我们着重介绍了新兴趋势和未来方向,为研究人员和临床医生提供了宝贵的见解,以推动BioLMs在日益复杂的生物学和临床应用中的发展。
1. 引言
大型语言模型(LLMs)的快速发展,如 BERT [1]、GPT [2] 及其专用版本,已彻底改变了自然语言处理(NLP)领域。这些模型在建模上下文、解析复杂数据模式以及生成类人响应方面的能力,自然而然地扩展到了生物信息学领域,其中生物序列的结构和复杂性常与人类语言相似 [3]。LLMs 已成功应用于基因组学(genomics)、蛋白质组学(proteomics)和药物发现等多个生物信息学领域,提供了传统计算方法难以企及的洞察 [4]。
尽管取得了显著进展,但在这些模型在生物信息学问题上的应用进行系统分类和全面评估方面,仍然面临挑战。鉴于生物信息学数据的多样性和生命活动的复杂性,研究这一领域常常充满困难,因为现有研究往往专注于有限范围的应用。这导致对 LLMs 在不同生物信息学子领域中广泛用途的理解存在空白 [5]。
本综述旨在通过全面概述 LLM 在生物信息学中的应用来应对这些挑战。通过聚焦生命活动的不同层面,本文从生命科学和生物医学应用两个主要视角收集并展示了相关研究成果。我们与领域专家合作,汇编了涵盖这些视角中关键领域的深入分析,例如核体分析(nucleoid analysis)、蛋白质结构和功能预测、基因组学、药物发现和疾病建模,包括在脑疾病和癌症中的应用,以及疫苗开发。
此外,我们提出了新术语 “生命活动因子”(Life Active Factors, LAFs),用于描述作为生命科学研究目标候选的分子和细胞组成。LAFs 的范畴极为广泛,不仅包括具体实体(如 DNA、RNA、蛋白质、基因、药物),还包括抽象成分(如生物通路、调控因子、基因网络、蛋白质相互作用)和生物测量(如表型(phenotypes)、疾病生物标志物(disease biomarkers))。LAFs 是一个综合术语,有助于协调跨越不同生物信息学子领域的概念差异,促进对 LAFs 多模态数据及其在复杂生物系统中相互作用的理解。LAFs 的引入契合基础模型的精神,强调 LAFs 的序列、结构和功能之间的统一性,同时尊重每个 LAF 作为生物网络中节点的相互关系。
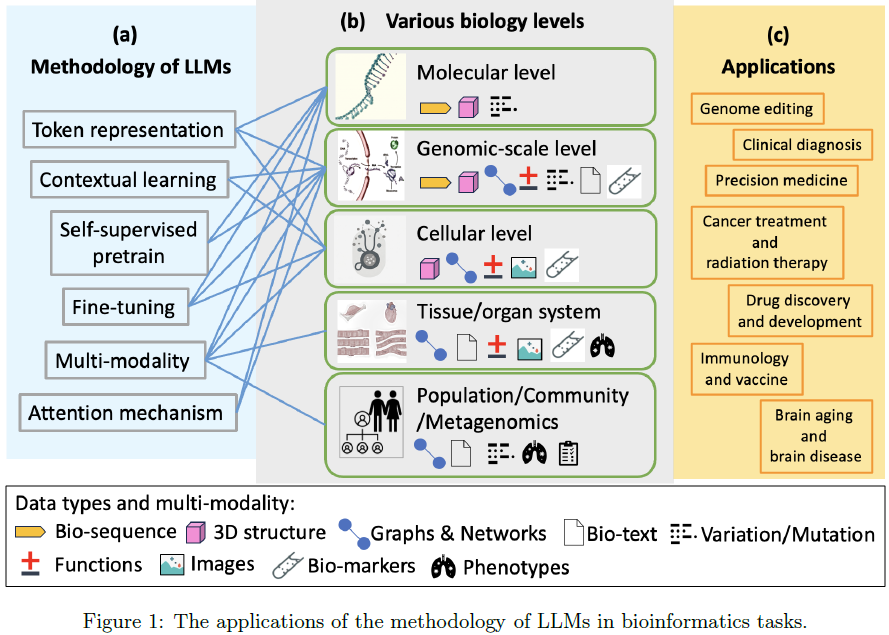
通过弥合现有知识的空白,本工作旨在使生物信息学家、生物学家、临床医生和计算研究人员能够理解如何有效利用 LLMs 解决生物信息学中的迫切问题。本综述不仅强调了近期的进展,还识别了尚未解决的挑战和机遇,为未来的跨学科合作与创新奠定基础(图 1)。
2. 生物信息学中语言模型与基础模型的背景
生物信息学已成为生命科学中一个基础性且变革性的领域,连接了计算技术与生物研究。该领域强调开发和应用计算工具与方法,以管理和解释大量的生物医学数据,将其转化为可操作的洞察,从而推动多个下游应用的进步。尤其是以深度学习技术为基础的现代计算工具,显著加速了生物学研究的演化。
LLMs 技术的快速进步为生物信息学计算带来了新的方法。鉴于生物系统的复杂性及生物信息学数据的高度结构化特性,基于 LLM 的计算方法已被证明能有效应对基因组学、蛋白质组学和分子生物学等领域的挑战。受 Transformer 等 LLM 架构的启发,生物信息学中的基础模型擅长捕捉生物数据中的复杂模式和关系。这些模型已经从单一模态工具演变为复杂的多模态系统,整合了基因组序列、蛋白质结构等多样化的数据集。
他们成功的关键在于大规模高质量训练数据的可用性,以及自监督预训练和微调技术的采用。这些方法使模型能够从无标签数据中提取有意义的特征,并适应特定的生物信息学任务。结合架构设计的进步,这些创新拓宽了基础模型的能力和影响力,解锁了对生物系统的新见解,并加速了生命科学领域的进展。以下部分将讨论这些先进的计算方法,以及生物系统的内在特性和结构化生物信息数据。
2.1 语言模型与生物信息学基础概述
2.1.1 LLM 与基础模型
传统语言模型旨在以类似人类的方式处理和生成文本,充分利用其训练期间使用的大量数据集。这些模型在解释上下文、生成连贯且符合上下文的响应、执行翻译、文本摘要以及回答问题等任务中表现卓越。大型语言模型(LLMs)是一种基础模型,它们通过庞大的数据集训练,提供灵活且强大的能力,用以应对广泛的应用场景和用例需求。这种统一的模型架构消除了为每个特定任务单独构建和训练领域特定模型的需求,从而克服了成本和资源限制。LLMs 不仅促进了任务间的协同作用,通常还能实现更优的性能,使其成为更具可扩展性和高效的解决方案。在适应生物信息学任务方面,语言模型的成功取决于几个关键因素(图 1(a))。
表征学习与分词(Representation learning and tokenization)。LLMs 的分词受到其分词算法设计的影响,主要使用基于子词级词汇的技术来有效地表示文本序列数据。常用的分词算法包括字节对编码(Byte-Pair Encoding, BPE)、WordPiece 和 Unigram 等。这些分词方法尽管无法完美捕捉输入表达的每种可能变体,但它们能够有效编码词汇的特征及其上下文关系。
在表征学习的角度上,语言模型的分词与词嵌入算法通常能够成功表征数据背后隐含的变化因素。这种表征基于语言模型的无监督学习模式。编码器模块或嵌入层中学习到的子词上下文特征遵循概率建模,并在大规模语料数据集中持续更新表征。
注意力机制(Attention mechanism)。LLMs 广泛采用 Transformer 模型作为其基础架构。Transformer 模型的核心创新是多头自注意力机制(multi-head self-attention),它能够在所有相关的标记(tokens)之间建立关系,从而更有效地编码输入序列中每个词的意义。
自注意力层处理一系列标记(类似于语言中的单词),并在整个序列中学习上下文信息。“多头”部分意味着多个注意力头可以同时运行,以捕获多样的上下文特征。在单个注意力头内部,序列中的一个标记输出嵌入与上下文中其他标记结合,通过合适的因果掩码实现全局信息融合。这种全局级别的注意力机制使得信息融合在可用的上下文窗口中更加高效。
自监督训练方法(Self-supervised training methods)。语言模型采用自监督学习方法进行训练。与通常需要人工标注的监督学习不同,语言模型能够利用大量未标注的文本数据。
无监督学习的目标是通过识别和捕捉未标注数据的有意义属性来进行分析。例如,自动编码器通过一个称为瓶颈层的隐藏层将数据压缩成低维表征,然后从该表征中重建原始输入数据。语言模型通过利用句子中的下一个词作为上下文的自然标签,或通过人工掩蔽(mask)已知词并预测它的方法(如预测下一个词或掩蔽词),利用非结构化数据生成标签,并以此进行自监督学习训练。基于 Transformer 的模型凭借其并行处理能力以及捕捉整个序列相关性的能力,达到了最先进的性能水平。
一种更高级的训练方式是文本到文本(text-to-text)框架。这种训练框架统一了包括翻译、问答、分类等多种任务,将其格式化并作为输入提供给模型,通过生成目标文本进行训练。这种被称为 “T5” 的框架受益于在多种任务中使用相同的模型、损失函数、超参数等。
预训练(Pre-training)。在许多监督学习问题中,输入数据由多种特征组成,包括数值或类别信息,这些特征可以帮助模型进行预测。从头开始训练的模型(scratch-trained models)需要从零初始化并使用任务特定数据集训练所有参数,通常需要大量迭代才能在单个任务上完全收敛。总体而言,基于 Transformer 的语言模型分为两类:从头训练的模型和预训练模型。LLMs 应用基于 Transformer 的预训练模型,这些模型通过大量未标注数据进行训练,然后针对特定任务进行微调。预训练从未标注数据中学习到一般信息,可以提高目标任务的收敛速度,并且比从头训练参数更具泛化能力。利用大规模语料库的上下文信息进行预训练(如整个模型或编码器模块),在多种下游任务中已实现最先进的结果。
2.1.2 生物信息学的应用与挑战
利用深度学习方法(如语言模型)解决生物信息学问题充满挑战。尽管深度学习模型在特定生物信息学应用(例如基因组学应用)中比最先进的(SOTA)方法表现出更高的准确性,并且擅长处理多模态和高度异质的数据,但仍然存在重大挑战。需要进一步努力以整合和分析深度学习所需的多样化数据集,用于基因组预测和预后任务。这对于开发可解释的语言模型尤为重要,因为这些模型能够识别新的生物标志物,并阐明不同生物学层次间的调控关系,例如病理条件下的组织和疾病状态。这些进展需要对复杂的生物信息学数据、相关任务及其相互关系有深入的理解。在本综述中,我们从两个视角讨论这些问题:不同的生物学层次及其固有的生命活动调控。
不同的生物学层次。虽然尚无统一的标准划分,但生物信息学中的生命科学因素层次可以从微观到宏观分为五个层次。以哺乳动物模式生物为模板,这些层次可以分为:分子层次、基因组规模层次、细胞层次、组织/器官系统层次,以及种群/群落/宏基因组层次(图 1(b))。
生物信息学通常专注于前三个层次(即分子层次、基因组规模层次和细胞层次)。
- 1)分子层次。分子层次分析目标是生物活性分子,包括核酸、氨基酸及其他小型生物活性分子,以及旨在解释该层次生命活动的相关实验。
- 2)基因组规模层次。基因组规模层次通过 DNA、RNA、蛋白质及代谢组学建模生命活动。该层次最著名的调控是 “中心法则”(The Central Dogma),揭示了亚细胞尺度主要生命活动因素之间的内在关系。整个亚细胞系统以分层方式建模,从 DNA、mRNA 和蛋白质开始,延伸至代谢组学(metabolomics),并最终推断表型(phenotype)。
- 3)细胞层次。理解细胞机制是生物学中的一个基本挑战,在生物医学领域尤其重要,特别是在疾病表型和精准医学相关研究中。以基因(DNA 中控制下游生命活动的特定核苷酸序列)为单位,基因的功能及其产物是这一层次的重要研究目标。构建一个全面、结构化、计算机可访问的基因功能及其变异表征对于理解细胞有机体或病毒的生物信息学至关重要。同时,基因网络及其产物的相互影响也是这一领域的一个挑战。单细胞测序技术允许我们在 mRNA 层次获取基因表达数据,为分析整个细胞系统奠定了基础。这些数据现已广泛用于识别发育过程中细胞状态、描述特定组织或器官以及评估患者特异性药物反应。
在本综述中,基因组和细胞层次的分子组成及其相关集合统称为生命活性因子(Life Active Factors, LAFs)。需要注意的是,序列表示格式是每种 LAF 最常见的观察形式。然而,多模态数据对 LAF 的表示也至关重要,例如高度结构化的数据格式用于记录功能描述、丰度、变异及表达情况。
固有的生命活动调控(Inherent regulations of life activities)。由于大多数生命活性因子(LAFs)在各生物学层次中以序列格式表示,基于 Transformer 的预训练语言模型特别适合分析这些序列。一个新兴的共识认为,这些序列体现了一种内在语言,可以通过语言模型解码。然而,为了在生命活动中发挥作用,单个 LAF 的核心逻辑是 “序列-结构-功能”。
以蛋白质组学分析为例,蛋白质序列可以被视为由氨基酸组成的字母串,与人类语言类似。最新的蛋白质语言模型利用这些格式化的字母表示来表征次级结构单元,这些单元组合形成负责特定功能的结构域。蛋白质语言模型还可以直接从一级序列推断完整的原子级蛋白质结构,并生成具有功能性的蛋白质,而这些蛋白质在进化过程中可能需要数亿年才能发现。
在生命活动中,LAFs 不仅存在不同层次间的重要调控关系,也存在层内关系。
1)基因组层次的调控。在基因组层次,基因通过调控 RNA 和蛋白质产物的生成来控制遗传性状。根据分子生物学的中心法则,DNA 中的基因被转录为信使 RNA(mRNA),然后翻译成基因产物(如蛋白质)。对于任意一种基因产物,无论是 RNA 还是蛋白质,都可以追溯到指导其合成的基因。这种可追溯性表明,全面理解基因的功能不仅需要考虑基因本身,还需要考虑所有相关产物的角色和功能。
基因之间会相互调控,并通过反馈回路形成基因调控网络中的循环依赖链条。在建模这些 “稳态” 基因的调控关系时,图神经网络风格的操作非常适用。在蛋白质层次,蛋白质之间也存在蛋白质-蛋白质相互作用(protein-protein interactions,PPI)。在信号通路(pathways)层次,整个系统可以看作一个超图,每个超边对应一个包括多个蛋白质的信号通路。
2)细胞层次的调控。在细胞层次,信号通路整合了单个基因或蛋白质产物,通过层内的相互调控完成特定的细胞功能。蛋白质通过多种方式相互作用,如抑制、激活或结合其他蛋白质,从而影响细胞内的表达水平或蛋白质丰度。这些相互作用被统称为 PPI。一些数据库通过注释功能性信息系统性地组织结果,例如利用基因本体(Gene Ontology, GO)注释信息,并使用基因组系统中分配给每个基因的唯一标识符(unique gene identifiers)。
2.2 训练方法与模型
2.2.1 预训练
预训练 是大型语言模型(LLMs)开发的关键阶段,模型通过在大规模、多样化的数据集上进行训练来学习基础语言表示。预训练通常采用自监督学习技术,例如掩码语言模型(如 BERT)或因果语言模型(如 GPT),使模型能够预测被掩盖的 token 或序列中的下一个单词。与传统深度神经网络(DNNs)通常在领域特定数据集(如 ImageNet )上进行预训练不同,LLMs 的预训练是在涵盖书籍、百科全书和网络内容等广泛领域的超大数据集上完成的。此外,LLMs 的预训练涉及拥有数十亿甚至数万亿参数的模型,与传统 DNNs 相比,其计算和资源需求显著更高。
- 预训练的 主要优势 在于模型能够在多种语言任务中实现广泛的泛化,经常无需额外的任务特定训练即可达到零样本或小样本性能。这种广泛的泛化能力使 LLMs 在涵盖自然语言理解、生成和推理的任务中表现出色。
- 然而,预训练的 劣势 包括较高的计算和能源成本,通常需要高性能硬件的分布式系统。此外,预训练模型可能会继承训练语料中的偏见和错误,导致生成的输出带有偏见或不理想的内容。
微调 是基于预训练模型的后续阶段,通过额外的监督或半监督训练使其适应特定任务或领域。此过程使用较小的、针对性的数据集优化模型以满足特定应用需求。
- 微调可分为任务特定微调(使模型专注于情感分析或机器翻译等特定任务);领域特定微调(为医学或法律等专业领域优化模型);以及指令微调(使模型以自然语言提示进行对齐响应)。
- 最近的参数高效微调方法,如 LoRA(低秩适配)和适配器,进一步提高了微调效率,通过仅更新模型的一部分参数,在保留预训练模型计算优势的同时优化了性能。
微调通过利用特定领域或任务的数据增强了模型在具体任务中的表现,在各种应用中实现了最先进的性能。然而,它也带来了挑战,如对微调数据集的过拟合风险,这可能削弱模型的泛化能力。此外,微调需要高质量的标注数据以确保在专业应用中的可靠性和准确性。
2.2.2 RLHF
基于人类反馈的强化学习(Reinforcement Learning with Human Feedback,RLHF)是大型语言模型训练流程中的关键附加阶段,旨在使模型输出与人类的偏好和期望对齐。预训练和微调赋予模型一般的语言理解和任务特定专业知识,而 RLHF 优化模型的行为,使其生成更符合人类价值观、指令或对话风格的响应,这在对用户交互质量要求较高的应用(如对话代理)中特别重要。
RLHF 包含三个主要组成部分:基于人类标注偏好的奖励模型、基于奖励模型优化模型行为的强化学习(RL)算法,以及用于完善奖励系统的迭代人类反馈。
- 1)奖励模型 通常通过收集人类评估者对模型输出的排序数据集来开发,这些排序数据集作为训练奖励模型的基础,用于预测给定输出的期望性。
- 2)强化学习算法,如近端策略优化(PPO),调整模型参数以最大化奖励模型预测的分数。
- 3)直接偏好优化(DPO)算法通过排序偏好数据集直接优化模型以优先选择更高排名的输出。
RLHF 的 主要优势 在于其能够使预训练和微调后的模型输出与人类期望对齐,提升连贯性、相关性和伦理合规性等特质。这种方法在缓解生成有害内容(如毒性、偏见或无关内容)方面特别有效。此外,RLHF 可以整合特定领域的人类专业知识,使模型更好地服务于小众应用。
然而,RLHF 也带来了一些 挑战。
- 首先,人类反馈的质量至关重要,设计不良的反馈机制或不一致的人类偏好可能导致模型行为次优甚至有害。
- 其次,RLHF 需要大量的人力标注资源以及计算成本高昂的强化学习训练。
- 此外,对奖励模型的过度优化可能导致模型利用奖励系统的漏洞而非真正改善输出——这一现象被称为 “奖励黑客”。
2.2.3 知识蒸馏
知识蒸馏(Knowledge Distillation,KD)已成为高效训练和部署大型语言模型(LLM)的关键方法,通过将嵌入在高容量教师模型中的知识转移到更小、更高效的学生模型中。本质上,学生模型学习模仿教师的预测结果和内部表示模式,从而显著减少了在预训练阶段的计算成本和内存需求。这一方法促进了更精简的 LLM 的开发,同时不牺牲其执行复杂语言任务的能力。
KD 的最新进展不仅仅局限于最终输出匹配。现代方法利用已建立的 LLM 生成的不仅是预测,还包括详细的推理步骤,这些通常被称为 思维链序列(chain-of-thought sequences)或 中间逻辑痕迹(intermediate logic traces)。这些丰富的注释随后可以纳入微调过程,使目标 LLM 能够在没有大量人工标注的情况下获得更深层次的问题解决能力并增强可解释性。通过整合这些推理路径,KD 不仅作为压缩机制,还将先进的批判性思维和推理能力传授给新训练的模型。
此外,最近的研究探索了将 KD 扩展到 支持专门或领域特定任务的应用场景,其中已建立的教师模型可以 引导目标 LLM 专注于与任务相关的知识,过滤掉不相关的信息。这一方法有助于生成与其预期应用更为契合的模型。此外,已有关于 KD 的贝叶斯视角,它提供了对其统计基础的透明解释,并为目标模型提供了强大的不确定性量化能力。
预训练、微调、KD 和 RLHF 的整合代表了 LLM 的全面训练范式。
- 预训练作为基础,通过大规模无监督学习为模型提供一般知识和语言能力。
- 微调使模型适应特定任务或领域,增强其在目标应用中的表现。
- KD 通过实现知识的转移,提高效率,
- RLHF 则优化模型的行为,使其与人类偏好对齐,确保输出既在功能上准确,又在社会上可接受。
这些阶段是互补的、迭代的。在 RLHF 过程中获得的洞察可以为微调数据集或方法的改进提供信息,而微调和 KD 的进展可以提升 RLHF 结果的质量。通过这一训练流程,不仅确保了 LLM 的强大和多功能性,还使其更易用并与以人为本的目标对齐。
这个多阶段训练范式在 OpenAI 的 ChatGPT 和 Anthropic 的 Claude 等最先进模型的开发中发挥了重要作用,并为未来领域的进展设定了基准。这些进展包括发布全规模和轻量版本,其中 KD 通常在优化后者方面发挥作用。
2.3 生物信息学特定数据集
2.4 模型演化与关键里程碑
生物信息学中 LLM 的发展标志着一段变革之旅。这些最初为自然语言处理任务开发的模型,如 BERT 和 GPT,在解决特定于生物信息学领域的挑战方面展现了巨大的潜力。借助其处理和生成序列的能力,LLM 已被应用于各种生物数据类型,包括 DNA、RNA、蛋白质和药物分子。
在基因组学领域,像 DNABERT 和 GROVER这样的模型通过对 DNA 序列进行训练,预测功能区域,如启动子和增强子(promoters and enhancers),并分析突变。同样,转录组学(transcriptomics)从 SpliceBERT 和 RNA-FM 等模型中受益,这些模型有助于理解 RNA 剪接和二级结构预测。对于蛋白质组学,PPLM 模型如 ProtTrans 和 ProtGPT2 增强了与蛋白质结构、功能和相互作用相关的预测。这些进展得益于基础的 transformer 架构,擅长处理顺序数据。通过微调这些预训练模型以适应领域特定任务,它们的实用性得到了扩展,应用于药物发现中,其中分子和蛋白质序列的 SMILES 表示被整合,用于预测相互作用和性质。
生物信息学中的一个显著突破是 AlphaFold 系列,它应用了先进的机器学习技术解决蛋白质结构预测的挑战。AlphaFold2(AF2)通过其前所未有的准确性,基于氨基酸序列预测蛋白质结构,彻底改变了结构生物学。其基于注意力的深度学习架构捕捉了复杂的蛋白质折叠模式,超越了传统的基于物理的和同源建模的方法。通过多序列比对(multiple sequence alignments,MSA)利用进化信息,AF2 提供了可靠的预测,即使在缺乏实验数据的情况下,也能显著减少获得蛋白质结构信息的时间和成本,加速了药物发现和功能基因组学的进展。
在 AF2 成功的基础上,AlphaFold3(AF3)引入了突破性的功能,尤其是在建模蛋白质复合物(包括蛋白质-肽相互作用)方面。从单一蛋白质结构预测到多组分生物装配体,AF3 解决了蛋白质-蛋白质对接和蛋白质-肽相互作用建模的挑战。通过其基于模板(template-based,TB)和无模板(template-free,TF)的方法,进一步扩展了 AlphaFold 系列的多功能性和影响力。
AlphaFold3的关键特性:
- 增强复杂结构的准确性:AF3在预测蛋白质-肽复合物结构方面表现优异,在具有挑战性的场景中实现了高百分比的准确模型;
- 创新的无模板建模:在保持模板预测优势的同时,AF3 引入了强大的无模板算法,即使在没有同源结构数据的情况下,也能生成可靠准确的多样化模型;
- 复杂的评分与排名:AF3 集成了先进的评分指标,如 DockQ 和 MolProbity,确保对预测结构的准确评估。其模型表现出更少的扭曲肽段或顺式非脯氨酸残基等问题,反映出改进的蛋白质类属性和几何质量。
从 AF2 到 AF3 的进展反映了计算方法的迭代优化,以应对日益复杂的生物学问题。虽然 AF2 侧重于单一蛋白质结构的预测,AF3 则强调生物系统内的动态相互作用,标志着向更加全面的分子生物学理解转变。这些创新强调了机器学习如何不断重新定义生物信息学,实现蛋白质结构和相互作用的准确、高效建模。AlphaFold 系列展示了生物学和医学领域变革性突破的潜力,为未来理解复杂生物系统的应用铺平了道路。
【注:接下来的几章是如图 1(c) 所示的一些具体应用,详见原论文】
7. 讨论与未来方向
尽管大型语言模型(LLM)在生物信息学中取得了显著成功,但仍面临诸多挑战。
数据。LLM 在生物信息学中的表现严重依赖于训练数据的质量,而现有的数据集,如基因组或蛋白质组序列,通常包含噪声和偏差。这个问题导致了不准确的预测和有限的泛化能力。此外,标注的生物学数据的有限可用性进一步妨碍了 LLM 适应多样化生物信息学任务的能力。
计算成本和可扩展性 是另一个重大挑战。LLM 是资源密集型的,在训练和推理时需要大量的计算能力和内存,尤其在分析超长序列(如跨越数千个碱基对的基因组区域)时,问题尤为突出。尽管基于 transformer 的架构取得了一些进展,但由于固有的内存限制,它们仍然难以有效地扩展到如此长的序列。
泛化能力和可解释性 仍然是关键问题。尽管 LLM 在特定任务上表现出色,但它们在未见数据集或任务上的泛化能力通常不足。此外,模型输出缺乏可解释性,使得研究人员难以理解背后的生物学机制,而这是验证结果所必需的。伦理和隐私问题进一步复杂化了 LLM 的应用,特别是在个性化医疗等敏感领域。使用患者数据训练模型引发了重要的伦理问题和潜在的隐私风险,限制了其广泛应用。
尽管面临这些挑战,LLM 在生物信息学中的未来仍然充满了激动人心的机遇。
- 未来的努力可能将集中在开发轻量高效的架构,如 LoRA 和 QLoRA,以减轻计算和内存需求。
- Transformer 变种和混合架构的创新预计将克服可扩展性问题,使得长序列生物信息学任务的分析更加有效。
- 整合多种生物数据类型,包括 DNA、RNA、蛋白质序列、表观遗传学和转录组学数据,将增强 LLM 生成全面生物学见解的能力。
- 改进可解释性也将成为优先事项,相关进展旨在可视化注意机制并揭示预测背后的生物学基础。
个性化医疗的应用凸显了 LLM 的变革潜力。例如,
- 它们可以通过根据个体患者量身定制治疗方案,预测药物疗效或基于基因组数据识别可能的副作用,从而彻底改变精准医疗。
- 通过开放数据倡议和跨学科合作应对数据稀缺问题,将进一步加速进展,使 LLM 在生物信息学中的应用更加广泛。
- 此外,随着 transformer 模型的逐渐成熟,探索替代架构可能会推动创新,超越当前的局限,确保该领域的持续进步。
这些趋势突显了 LLM 在生物信息学中的动态演化,既带来了突破性发展的机会,也强调了必须解决现有局限性的需求。
整合 多模态生物医学数据 为未来研究提供了另一个有前景的方向。
- 序列到序列(Sequence-to-sequence)模型在自然语言处理中的显著成功,提供了一个有前景的技术方法来融合多种生物医学数据类型。这些模型可以潜在地弥合不同模态之间的差距——包括医学影像、临床文本、时间序列数据(如电子健康记录和生命体征)以及各种形式的生物序列数据(DNA、RNA和蛋白质)。例如,序列到序列架构可以在模态之间进行迁移,例如将放射学图像转换为诊断文本描述,同时结合相关的基因组信息。这种多模态融合可以通过利用来自不同数据源的互补信息,提供更全面的疾病诊断和治疗规划。
- 此外,创新的注意机制和跨模态 transformer 可能有助于捕捉不同数据类型之间的复杂关系,从而导致更强大且可解释的模型。挑战在于开发能够有效处理这些数据类型固有异质性的架构,同时保持计算效率和生物学可解释性。
8. 结论
本综合性综述探讨了LLM在生物信息学中的变革性影响,涵盖了基因组学、蛋白质组学、药物发现和临床医学等应用。我们的回顾强调了 transformer 架构在生物序列中的成功适用,专业化生物医学 LLM 的出现以及多模态数据整合的前景。这些进展推动了蛋白质结构预测、药物靶点相互作用分析和疾病诊断方面的重大进展。
尽管取得了显著成就,数据质量、计算可扩展性、模型可解释性和患者隐私等伦理问题仍然存在挑战。这些挑战为未来的研究提供了机会,特别是在开发高效架构、改善多模态数据整合和确保模型可解释性方面。LLM 与新兴生物技术的融合有望加速生物信息学的发现,可能带来更精确和个性化的医学干预。
论文地址:https://arxiv.org/abs/2501.06271
进 Q 学术交流群:922230617