BIOINFORMATICS期刊目前被中科院分为三区期刊,但是这个期刊的文章水平还是不错的,很适合打基础的同学或者跨学科的同学先拿来作入门期刊。上面当然也有一些比较复杂的文章,需要一些深度学习和生物相关的基础知识才能读懂(比如异质网络、各种信息矩阵的内容)。
一.MFR-DTA: a multi-functional and robust model for predicting drug–target binding affinity and region
用于预测药物-靶标结合亲和力和区域的多功能稳健模型 2023.2 三区
问题:一维卷积和 MLP 都完全忽略了每个元素的单个特征;LSTM 和 GNN 直接提取单个特征,但不足以获取全局特征;2D 卷积通过增加卷积核或堆叠更多卷积层来提取单个和全局特征,但其计算消耗却在快速增长;基于Transformer的骨架在参数上过于冗余,使其对高效的蛋白质和药物特征提取不太友好;结合位点的准确性难以确定
1.模型
作者设计了一种新的生物序列特征提取模块,即BioMLP/CNN,它帮助模型提取序列元素的单个特征,并且提出了一个新的Elem特征融合块来细化提取的特征。之后,构建了一个混合解码器模块,该模块提取药物-靶标相互作用信息并同时预测其结合区域。最后,提出了一个新的数据集sc-PDB,以更好地测量结合区域预测的准确性。
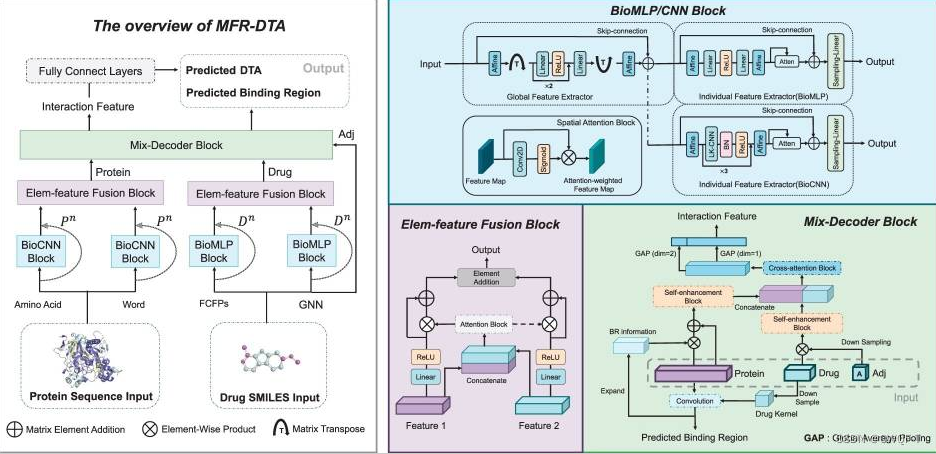
1.生物序列特征提取模块,即BioMLP/CNN,它包含一个全局特征提取器和一个单独的特征提取器,其输入是药物或蛋白质特征表示(参考了论文CPInformer的输入)。首先使用全局特征提取器来提取不同序列的相关性Xout,然后再使用两个单独的特征提取器来进一步操作Xout来得到组成元素的单个特征。ATT()操作使用空间注意力块来捕捉相邻元素之间的局部关系,进一步丰富提取的单个特征。有残差连接。
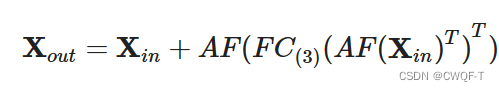
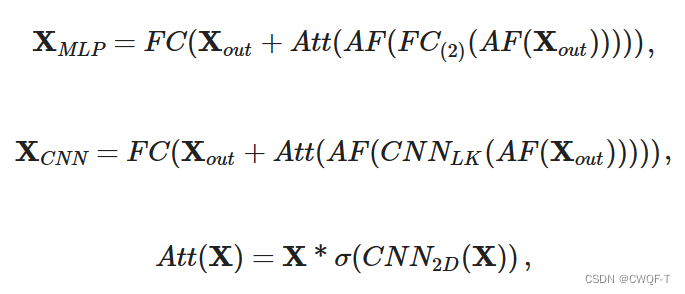
2.Elem-特征融合块:Wf大小是L*Cs,Cs是BioMLP/CNN模块提取的药物和蛋白质特征的通道数
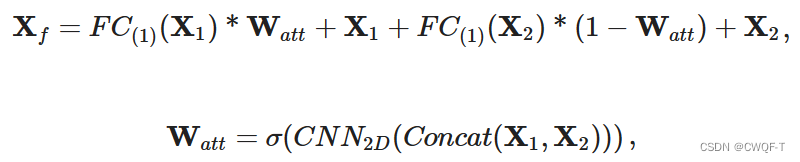
3.混合解码器模块:Mix-Decoder可以同时预测结合区域并且提取交互特征。输入是三个矩阵:药物特征矩阵、蛋白质特征矩阵、药物邻接矩阵Adj。
预测结合区域的过程如下:首先通过线性层对药物特征进行采样得到药物Kernal——Kd(大小是Cs*Cs),然后用Kd来过滤蛋白质特征Fp得到药物靶标反应向量S。在响应向量中,具有最高值的元素被标识为结合区域。(*代表逐元素相乘)
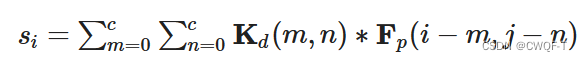
获取DTI特征步骤如下:首先通过重复来将s扩展为矩阵大小,再与蛋白质特征相乘以突出结合区域。对于药物,先通过全剧平均池化下采样Adj矩阵变成一个原子连接向量,再把这个向量通过重复来扩展成Adj信息矩阵Mc。两个矩阵通过自增强模块得到增强之后的特征矩阵。
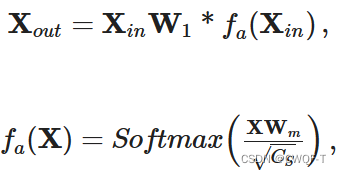
再把增强之后的特征矩阵连接起来,通过一个交叉注意力模块得到交互特征之后,再在一、二度上分别进行全局平均池化,最后采样并且连接起来之后得到最终的交互特征。
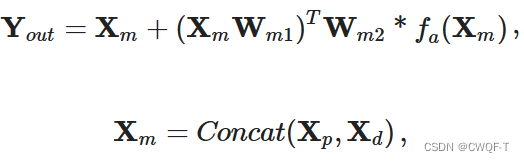
特别地,采用RWing损失函数。
1.药物表示
FCFP指纹和GNN分子图特征
2.蛋白质表示
使用氨基酸嵌入 (AAE) 和词嵌入 (WE)
2.实验
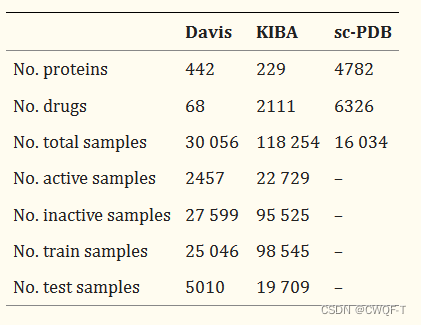
1.数据集
KIBA、DAVIS,sc-PDB
3.结果
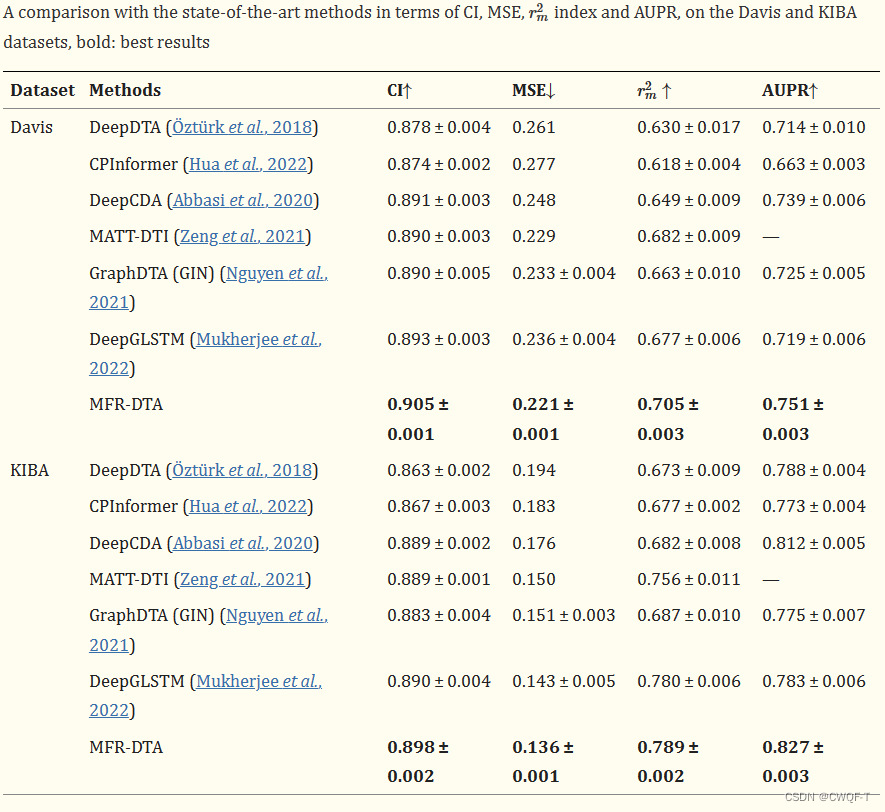
还进行了一系列消融实验:使用CPInformer作为基线,分别添加上述三个模块来作为改进,还用 Mix-Decoder 模块替换了基线方法中的原始 ProbSparse 自注意力模块,故一共做了四个消融实验。因为涉及到其他模型,故此处先不做讲解。这篇论文的操作比较复杂,需要深扒一下代码。
二.DeepDTA: deep drug–target binding affinity prediction
药物-靶标结合亲和力预测 2018.9 三区
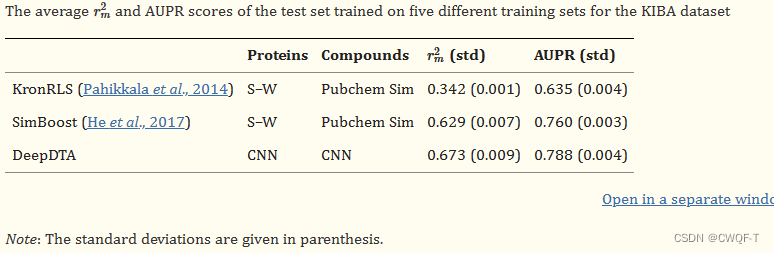
这是一篇药物靶标亲和力领域经常被比较的一种方法,虽然目前看来所使用的方法比较老,但是有很大的意义。作者使用CNN来学习蛋白质的表征,并使用配体的预定义Pubchem Sim评分。使用这种组合并没有改善结果,这表明使用CNN架构不足以有效地从氨基酸序列中学习。但是用CNN处理药物和蛋白质,得到的结果比较好,所以就用CNN处理蛋白质了。
1.模型
该模型由两个独立的CNN块组成,每个块都旨在从SMILES字符串和蛋白质序列中学习表征。对于每个 CNN 块,使用了三个连续的 1D 卷积层,filter数量不断增加。第二层的filter数量是第一层的两倍,第三层的filter数量是第一层的三倍(最后每个 CNN 块由 32、64、96 个滤波器的三个一维卷积组成)。卷积层之后是最大池化层。最大池化层的最终特征被连接起来,并被输入到三个FC层中。在前两个 FC 层中使用了 1024 个节点,每个节点后跟一个比例为 0.1 的dropout layer。第三层由 512 个节点组成,然后是输出层。
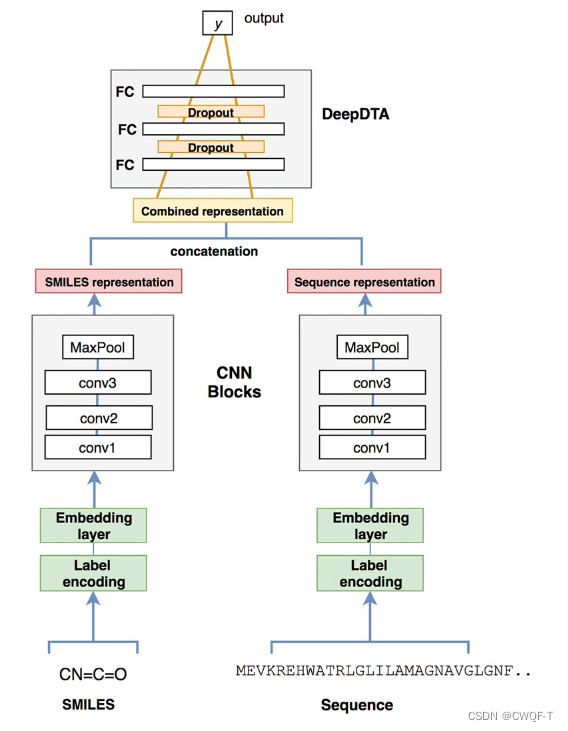
1.药物表示
SMILES,把整数和每个字符进行映射。KIBA数据集最大100个字符长度,Davis数据集最大85个字符长度。短的补充0,长的截断
2.蛋白质表示
氨基酸序列,把整数和每个字符进行映射。KIBA数据集最大1000个字符长度,Davis数据集最大1200个字符长度。短的补充0,长的截断
2.实验
1.数据集
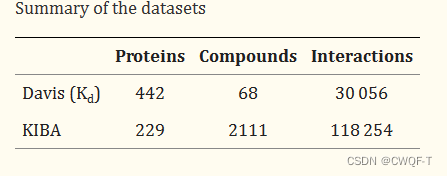
将数据集随机分为六个相等的部分,其中一部分被选为独立测试集。其余的进行五折交叉验证。
3.结果
S-W表示:用Smith-Waterman算法构建蛋白质相似性矩阵。Pubchem Sim表示使用 Pubchem 结构聚类服务器 (Pubchem Sim) 计算SMILES的相似性矩阵
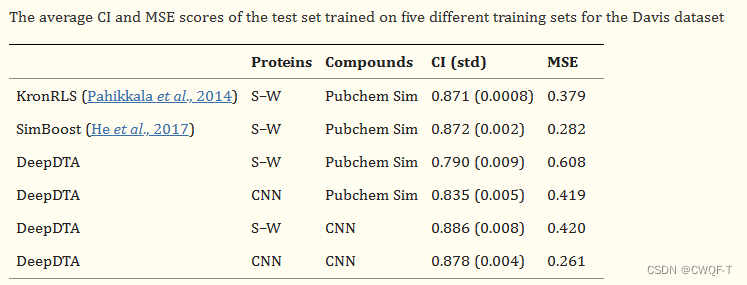
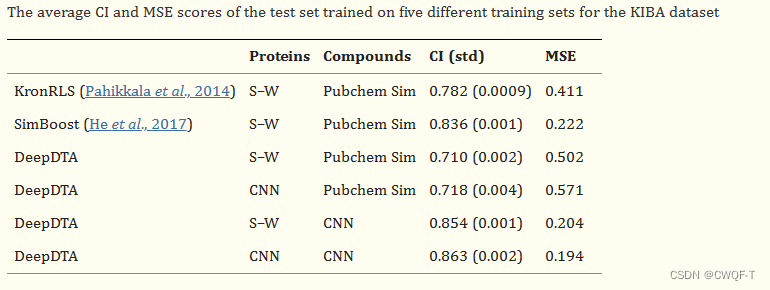
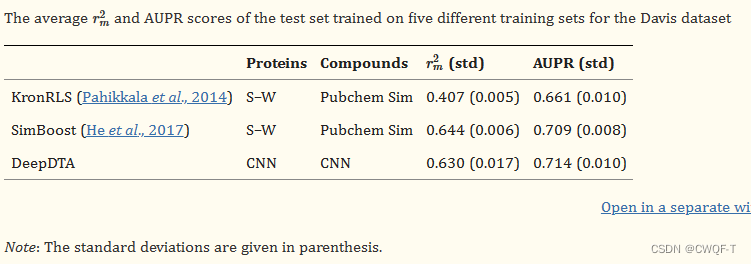
还使用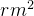
三.GDilatedDTA: Graph dilation convolution strategy for drug target binding affinity prediction
用于药物靶标结合亲和力预测的图扩张卷积策略 2024.2 二区
1.模型
包括三个模块:特征编码模块(FFM)、表示学习模块(RLM)、DTA预测模块(DTAPM)。
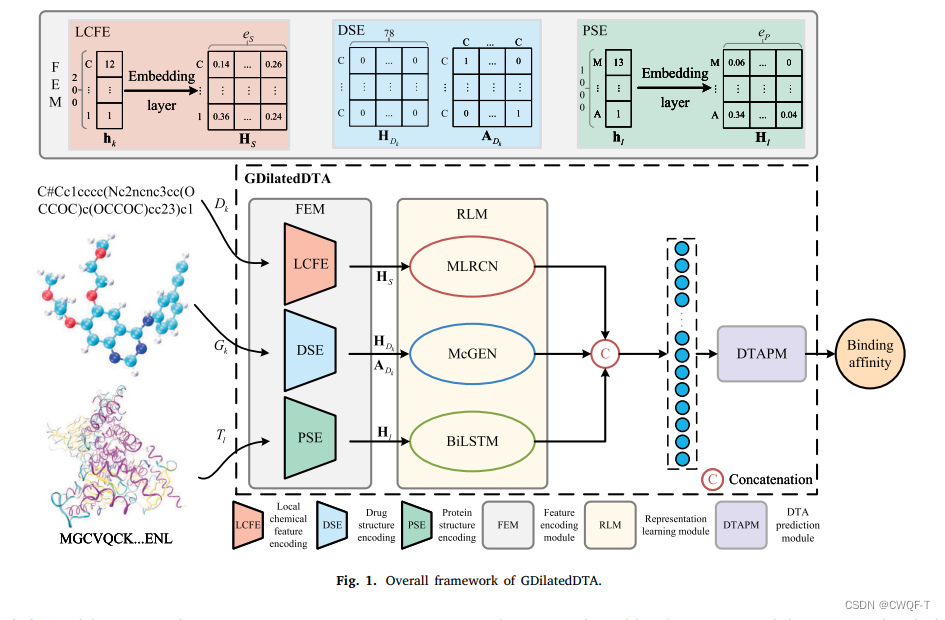
1.FEM 对药物-靶标对的初始特征信息进行编码:使用三个独立的组件对药物-靶标对的初始特征信息进行编码,即局部化学特征编码(LCFE)、药物结构编码(DSE)和蛋白质结构编码(PSE)。
(1)局部化学特征编码:构建数字和字符之间一对一映射的关系。药物长度设置为200,长的截断,短的补0。所以嵌入之后就成了200*Hs大小。Hs是特征向量的维度。
(2)药物结构编码:将SMILES转化为分子无向图,利用78维二进制特征向量表示药物。78维特征向量包含五种类型的信息,包括原子符号,相邻原子数,相邻氢原子数,原子隐含化合价,以及原子是否属于芳烃结构。

(3)蛋白质结构编码:设置最大长度为1000,短的补0,长的截断。最终蛋白质嵌入表示为1000*Hp大小的矩阵。Hp表示每个氨基酸的特征向量的维度。
2.RLM使用不同的组件来获得药物和靶点的潜在特征表示:包括 MLRCN 模块、McGEN 模块和 BiLSTM 模块三个组件。
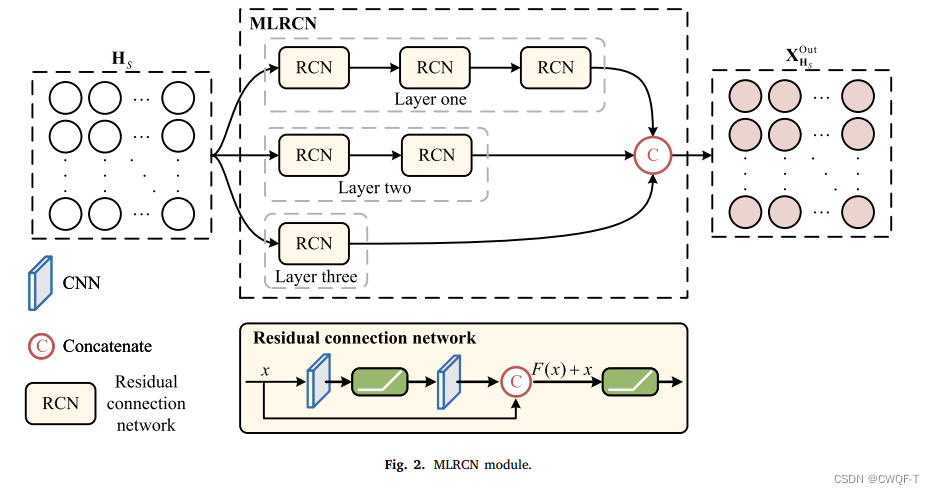
(1)MLRCN 模块:利用残余连接网络(RCN)实现从 SMILES 序列中提取局部化学特征信息。每个RCN内包括两个CNN层,后跟一个ReLU激活函数。MLRCN 由三层组成,其中第一层有 3 个 RCN 层,第二层有 2 个 RCN 层,第三层有 1 个 RCN 层。第一层中每个RCN包括128 和 96 个 CNN 节点(应该是filter的大小?),第二层包括 96 个和 64 个,第三层包括 64 个和 32 个。
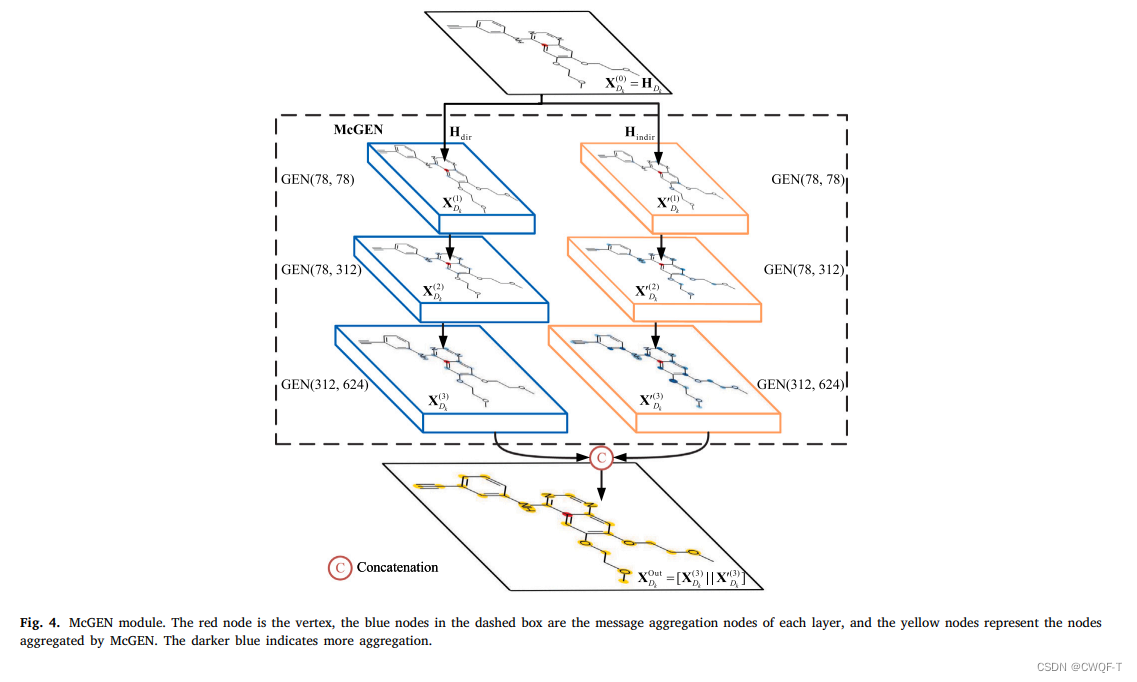
(2)McGEN利用通用聚集网络GEN(论文Improving graph neural network expressivity via subgraph isomorphism counting提出)组成药物分子图全局结构特征的双通道架构。每个通道包含三个GEN。GEN(78, 312) 表示输入和输出的特征通道数分别为 78 和 312。
(3)BiLSTM:氨基酸序列特征从两个方向输入到LSTM模块中,然后BiLSTM组合两组特征并输出目标的嵌入特征表示。
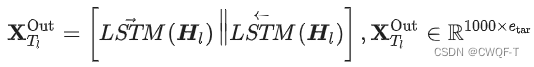
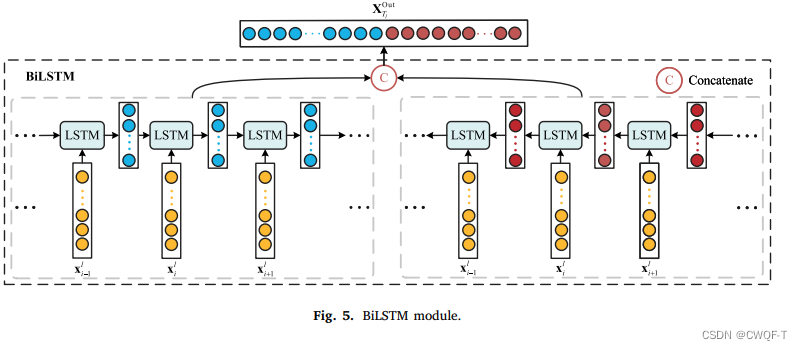
3.DTAPM 合并这些特征并预测它们之间的亲和力:
DTA prediction module将上述三个模块的输出拼接起来作为输入数据进行预测。
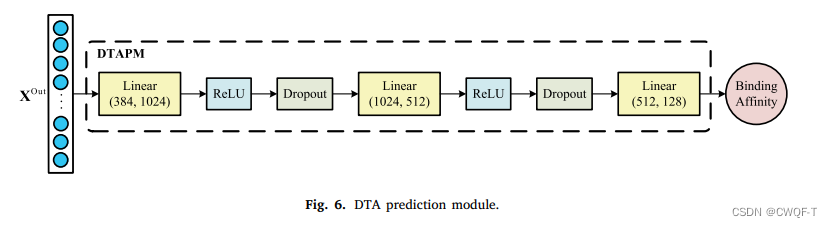
1.药物表示
SMILES和分子图
2.蛋白质表示
氨基酸序列
2.实验
1.数据集
Davis数据集包含 442 种蛋白质、68 种药物分子以及由这些蛋白质和药物组成的 30056 个相互作用对。该数据集中药物和蛋白质之间的结合亲和力通过解离常数来衡量Kd.较小的Kd值表示较强的亲和力。变化为对数之后,用于表征结合亲和力的范围是[5,10.8]。
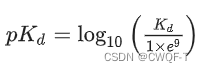
KIBA数据集包含 229 个蛋白质靶点、2111 个化学小分子和 118254 个药物-靶点相互作用对。该数据集中的 DTA 由 KIBA 评分衡量,该评分由半抑制浓度IC50和抑制常数Ki和Kd组合计算得出。
3.结果
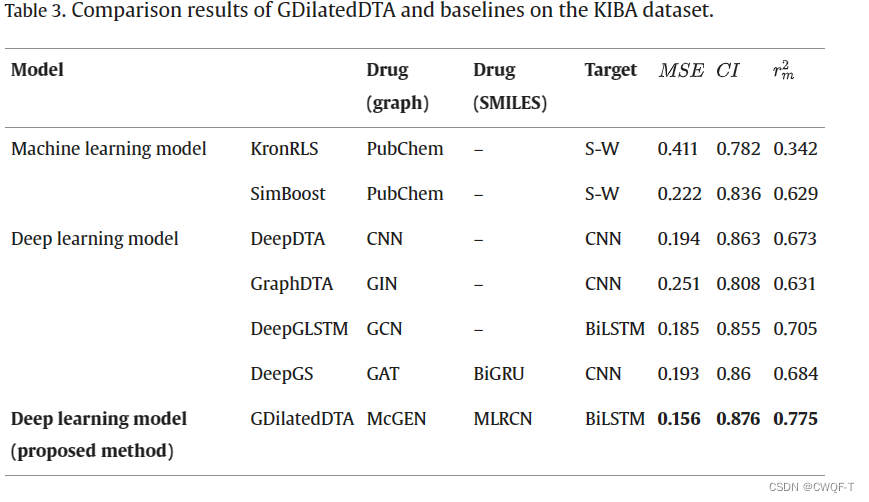
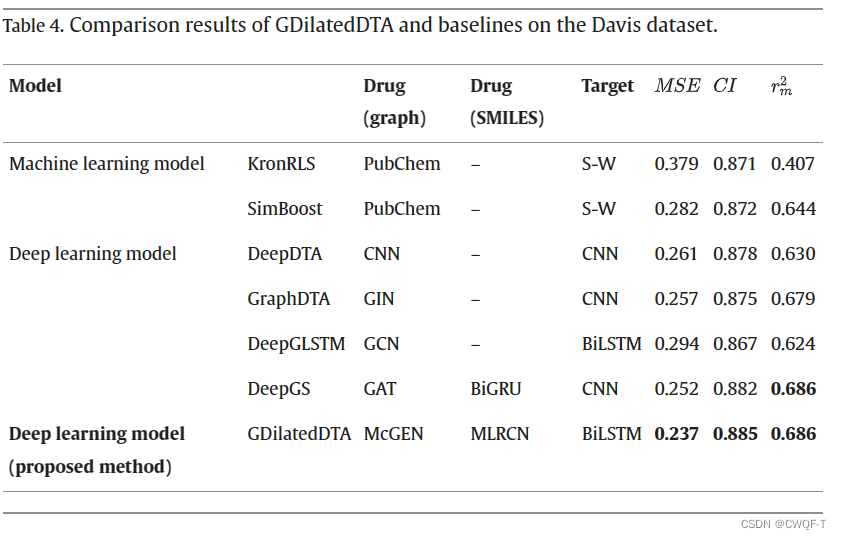
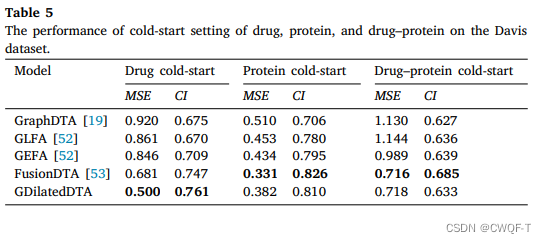
当在训练集中没有出现一些数据的时候,评估模型在面对新环境(例如突变蛋白质)时的鲁棒性。
四.DeepGLSTM: Deep graph convolutional network and LSTM based approach for predicting drug-target binding affinity
基于深度图卷积网络和LSTM的药物-靶标结合亲和力预测方法 2022.1 被录用在2022 SIAM International Conference on Data Mining (SDM)论文集中
1.模型
使用GCN处理药物,使用BiLSTM处理蛋白质。
1.对于药物来说,节点特征被表示为多维二进制向量,包括五种信息:包括原子符号,相邻原子数,相邻氢原子数,原子隐含化合价,以及原子是否属于芳烃结构。模型包含三个GCN模块:第一个包含三个GCN层,第二个包含两个,第三个包含一个。
2.对于蛋白质来说,将每种氨基酸与英文字母一一映射来进行嵌入,再传递给BiLSTM。得到正反两个方向的向量进行拼接,输出得到最终的特征向量
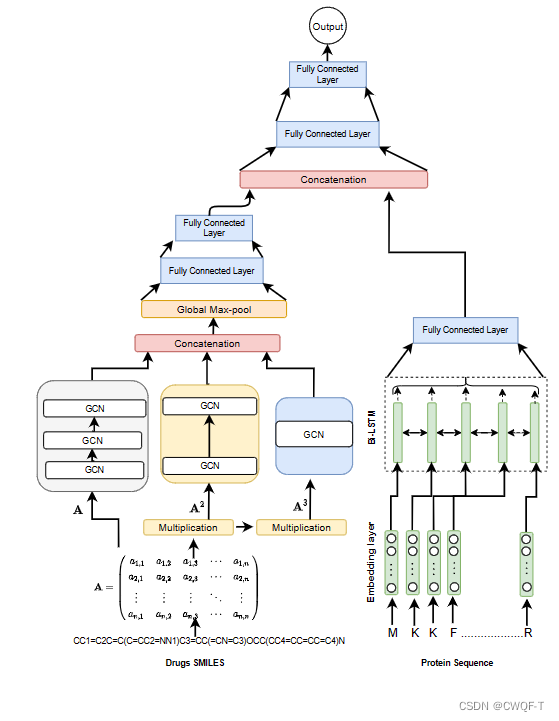
1.药物表示
分子图
2.蛋白质表示
氨基酸序列
2.实验
1.数据集
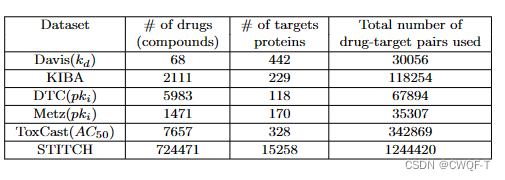
3.结果
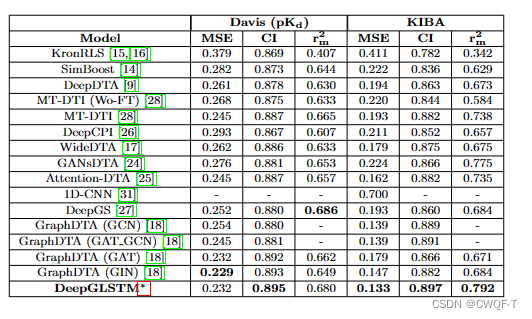
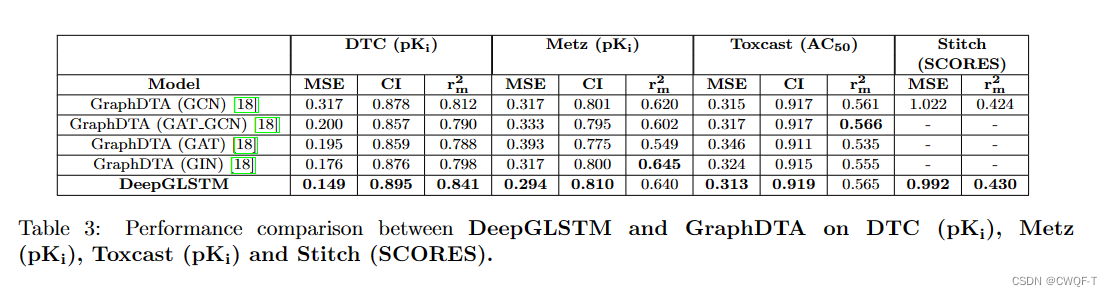
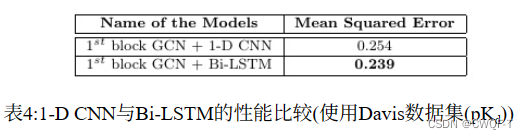
消融实验:使用CNN处理蛋白质序列