概述
在生命科学的“造物革命”中,蛋白质工程一直面临着“试错成本”与“设计效率”的双重挑战——传统方法依赖繁复的多序列比对(MSA)或耗时的实验室筛选,如同在浩瀚的蛋白质宇宙中盲选星辰。而今日,一项发表于《Cell Research》的突破性研究彻底改写了游戏规则:中国科学家团队开发的ProMEP(Protein Mutational Effect Predictor)通过多模态深度学习,仅凭单条蛋白质序列与预测结构,即可实现零样本突变效应预测,无需MSA辅助,将基因编辑工具TadA的A-to-G转化效率推至77.27%,同时使TnpB核酸酶的编辑效率提升近3倍!这项技术不仅比传统方法快数百倍,更首次证明AI模型通过整合1.6亿蛋白质的序列与结构信息,能精准预测人类从未见过的蛋白质突变效果,为“按需设计生命元件”按下加速键。
这篇论文的题目是《Zero-shot prediction of mutation effects with multimodal deep representation learning guides protein engineering》论文链接
下面我对这篇论文进行结构化介绍,帮助一下读者快速掌握这篇论文核心。
我写了一篇赛博修仙版,搭配食用效果更佳:
 https://blog.csdn.net/weixin_47520540/article/details/145813189?sharetype=blogdetail&sharerId=145813189&sharerefer=PC&sharesource=weixin_47520540&spm=1011.2480.3001.8118
https://blog.csdn.net/weixin_47520540/article/details/145813189?sharetype=blogdetail&sharerId=145813189&sharerefer=PC&sharesource=weixin_47520540&spm=1011.2480.3001.8118论文解剖指南:把天书拆成乐高积木
在正式开箱ProMEP这个"蛋白质预言家"之前,请允许我祭出科研江湖生存指南——当年在本科实验室摸鱼时认真学习时,导师传授的论文六脉神剑:
"看东西就关注三点:数据、输入、输出;训练方式、度量方法(loss)、评价体系;网络和特殊设计"
弟子不才,对其进行一下转述:"看AI论文就像做菜:食材(数据)决定上限,菜谱(网络结构)决定下限,火候(训练策略)决定成败,最后还得靠米其林评委(评价指标)盖章认证。"
一、数据部分——蛋白质宇宙的「灵气源泉」
主要让大家看看AI里面的蛋白质数据长啥样,有个基本的把握(可不是一个大分子结构团哦,AI模型可吃不下)
(一)基因编辑酶TnpB和TadA中氨基酸的变异位置的概率
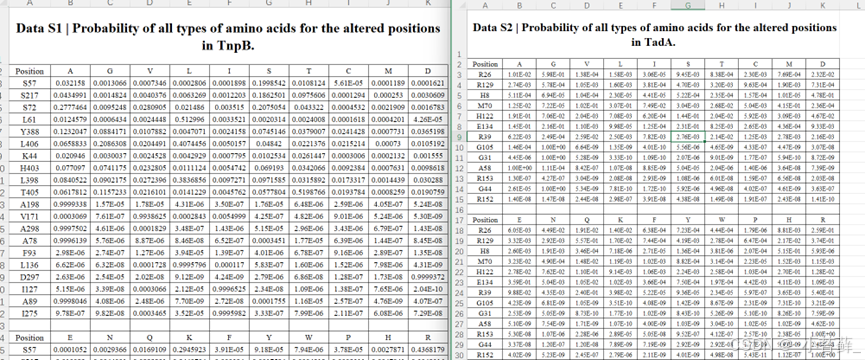
图Source_Data1 AAProbability-score-TnpB.xlsx 和 Source_Data2 AAProbability-score-TadA.xlsx
注:所有图片来自论文开源的数据集
- Position:表示氨基酸在蛋白质序列中的位置(即该氨基酸在序列中的序号)。
- Wild-Type Amino Acid:表示在该位置上的天然氨基酸(即未突变前的氨基酸)。
- Mutated Amino Acid:表示突变后的氨基酸(替换了天然氨基酸的氨基酸)。
- Probability Score:表示模型预测的突变后蛋白质适应性分数,这个分数越高,表明突变后的氨基酸在该位置上更可能维持或增强蛋白质功能。
-
具体介绍:每一行代表蛋白质序列中的一个具体位置及其相应的突变信息,包括该位置上的天然氨基酸、可能的突变氨基酸,以及模型为此突变计算出的适应性概率分数。
这些数据用于评估模型预测的准确性。模型在训练过程中学习如何根据序列和结构信息来预测突变的适应性分数,并通过这些分数来指导蛋白质工程(例如,识别出能够提高酶活性或稳定性的有益突变)。
-
(二)TadA中40个有益突变的编辑效率。
-
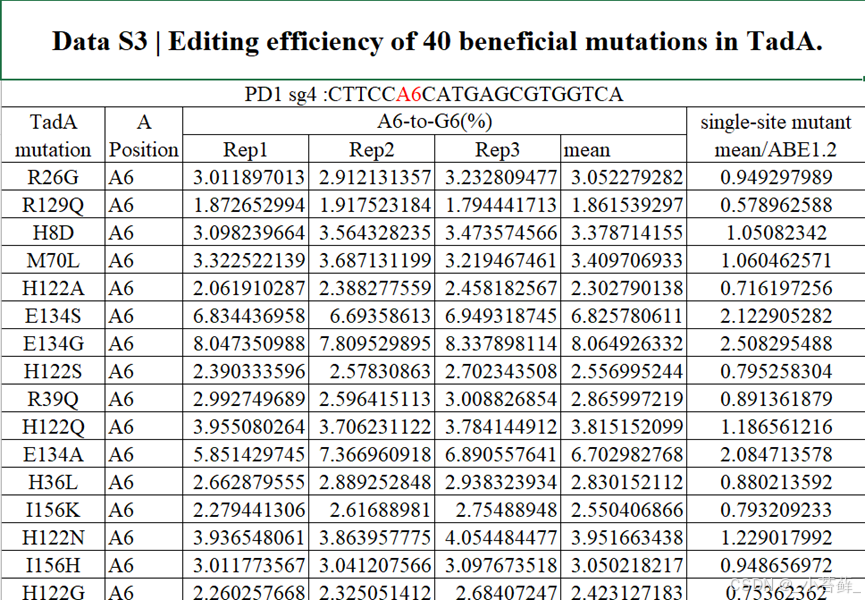
Source_Data3-6.xlsx
- Position:与上面表格类似,表示蛋白质序列中的氨基酸位置。
- Wild-Type Amino Acid:表示在该位置的天然氨基酸。
- Mutated Amino Acid:表示突变后的氨基酸。
- Probability Score:表示模型对每个突变后蛋白质功能的预测分数。
- Additional Columns:可能包含多个额外信息列,如不同突变组合的适应性分数、实验测量值等。
具体:每一行对应一个特定的突变组合及其相关的适应性预测分数。多个突变可能会组合在一起,以显示这些组合对蛋白质功能的影响。
该表格的数据帮助模型学习如何处理复杂的多点突变情景,尤其是涉及多个氨基酸位置同时发生变化的情况。这些数据提供了实际生物实验的参考,以验证模型预测的可靠性和实用性。
二、模型输入
模型的输入是多模态的,主要包括蛋白质的序列信息和结构信息。
1、蛋白质序列
简单来说就是由氨基酸按照特定顺序组成的链,接收一个蛋白质的氨基酸序列,例如"MKVLYNLVNA..."(序列输入首先通过一个嵌入层(embedding layer)进行编码,这个层将每个氨基酸转换成一个向量(矢量),这些向量捕捉了氨基酸的物理化学性质以及它们在蛋白质中的上下文关系。每个氨基酸的嵌入通常是一个高维的向量,比如128维或更高维度的向量,这样可以更全面地表示其性质。)
2、蛋白质结构输入
涉及到蛋白质的三维构象,即蛋白质中各个原子的位置和它们之间的空间关系。
- 蛋白质点云:模型采用了一种名为“蛋白质点云”的表示方法。蛋白质点云是一组三维坐标点,每个点代表一个氨基酸的α碳原子(即该氨基酸的主链中的一个关键原子)。这些点不仅有空间坐标(x, y, z),还附带了该氨基酸的类型(如G, A, V等)和在序列中的位置。
- 输入形式:这些点云数据通过模型的结构嵌入模块进行处理。模型利用这些三维坐标和氨基酸类型来捕捉蛋白质的空间构型和氨基酸之间的相互作用。
3、组合输入
在多模态模型中,序列信息和结构信息并不是独立处理的,而是通过特定的架构(如编码器-解码器架构)结合在一起,以便模型能够同时理解和处理蛋白质的线性序列和三维结构。
- 序列上下文:模型的序列嵌入模块使用Transformer架构,能够捕捉序列中的长程依赖关系和氨基酸之间的复杂相互作用。
- 结构上下文:模型的结构嵌入模块(如SE(3)-Transformer)则能够处理蛋白质的三维信息,确保模型能够识别蛋白质中空间上相互靠近但在序列上可能相隔很远的氨基酸之间的相互作用。
三、模型输出
模型的输出为每个突变体的适应性预测分数,这些分数表示突变后蛋白质功能可能发生的变化(如活性增加或减少)。此外,模型还能够预测多点突变的综合效应,以帮助识别具有潜在有益功能的突变组合。
四、训练方式
训练数据
模型在AlphaFold2数据库中预测的约1.6亿个蛋白质结构上进行自监督训练。训练数据包括从这些蛋白质中提取的序列和结构信息。
训练方法
模型采用了自监督学习的方式进行训练,这意味着模型在训练过程中不需要人工标注的数据,而是通过掩码预测来学习数据的内在结构。(这里补充一下掩码策略)
掩码策略:
- 序列掩码:在输入的蛋白质序列中,随机选择15%的氨基酸进行掩码。被掩码的氨基酸有80%的概率被替换为一个特殊的掩码标记,有10%的概率被替换为随机的另一个氨基酸,剩下的10%保持不变。模型的任务是根据上下文信息预测这些掩码处的真实氨基酸。
- 结构掩码:对于蛋白质的点云结构,模型会掩码掉靠近蛋白质中心的25%的点,然后通过结构信息来重建这些点的三维坐标。
五、度量方法
损失函数
交叉熵损失(Categorical Cross-Entropy, CE):用于评估模型预测的突变氨基酸与实际氨基酸之间的差异,主要用于序列重建。
Chamfer距离损失(Chamfer Distance, CD):用于度量重构后的蛋白质点云与真实结构之间的几何差异,确保模型能够准确捕捉蛋白质的三维结构信息。
六、评价指标
斯皮尔曼等级相关系数(Spearman’s Rank Correlation):用来评估模型预测结果与实验测量之间的相关性,适用于无监督预测任务。(关于这个指标我的这篇博客中有介绍:斯皮尔曼相关系数)
受试者操作特性曲线下面积(Area Under the ROC Curve, AUROC):用于评估模型在区分病原性突变和非病原性突变方面的表现,特别是在病原性预测任务中使用。
平均精确度(Mean Average Precision, MAP):用于多任务预测中的精度评估。
七、模型设计——压轴大戏
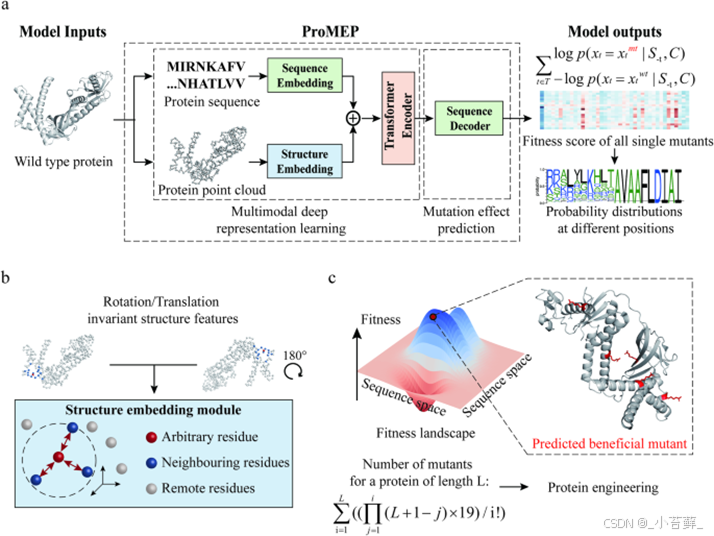
a:以任意 WT 蛋白质作为输入,具体而言,对于任意突变,ProMEP 首先从 WT 蛋白质中提取序列嵌入和结构嵌入。然后对这些嵌入进行对齐并输入到预训练的 Transformer 编码器中,以生成残差分辨率的蛋白质表示。使用序列解码器,细粒度蛋白质表示最终分解为序列和结构背景下每个氨基酸的条件概率。任意突变的影响可以解释为突变序列和 WT 序列之间预测对数似然的差异。采用定制的蛋白质点云以原子分辨率引入蛋白质结构背景。
b:输入蛋白质结构的 3D 平移和旋转不会影响蛋白质的结构背景。 ProMEP 应用旋转和平移等变结构嵌入模块来保证这种不变性。
c :ProMEP可用于指导蛋白质工程,而无需标记数据集或对蛋白质结构和分子功能的整体理解。它使用户能够通过有效遍历蛋白质适应度景观来识别有益的(多个)突变体。
模型结构
1、多模态深度学习模型:该模型结合了蛋白质序列和结构的多模态信息,通过编码器-解码器架构学习蛋白质的序列和结构上下文。编码器负责处理输入的掩码序列和点云数据,生成特征表示;解码器则用于重建掩盖的信息。
2、Transformer编码器:由33层堆叠的Transformer组成,每层包括层归一化、20头注意力块和前馈网络,用于捕捉序列信息的上下文。
3、SE(3)-Transformer结构嵌入模块:保证结构上下文在三维变换中的不变性,确保模型对输入结构的旋转和平移具有不变性。
关键设计
- 蛋白质点云:使用蛋白质结构的α碳原子坐标构建点云,保持了蛋白质的几何信息,同时提高了计算效率。
- 多模态训练:同时学习蛋白质序列和结构上下文,确保模型能够整合多种信息来源,从而提供更准确的突变效果预测。
总结展望(科技狂想症犯了)
ProMEP虽强,但科学家的脑洞永远比AI大——这些升级方向正在路上:
🔥 挑战1.0:插入/删除突变
当前模型像精准的「氨基酸狙击枪」,但面对插入或缺失(InDels)这类「霰弹枪式改造」仍力不从心。解法?把训练目标从填空游戏(MLM)切换成接龙预测(Next Token),不过需要更庞大的算力和数据燃料!
🚀 挑战2.0:超长蛋白的「分块处理」
遇到新冠刺突蛋白这类「基因长篇小说」,ProMEP得像读PDF一样拆分成段落分析。未来可能用循环记忆Transformer实现「无限滚动阅读」,彻底告别上下文限制。
🤝 挑战3.0:蛋白质社交网络
现在ProMEP专注「单身蛋白」,若能整合蛋白质相互作用(PPI)数据,就能分析「蛋白复合体派对」——这对药物靶点设计简直是降维打击!
💡 未来科技树点法
-
强化学习(RL):让AI化身「突变策略师」,通过试错奖励机制自动优化设计路线
-
生成对抗网络(GANs):生成海量虚拟突变体,帮模型突破数据局限
-
图神经网络(GNNs):把蛋白质结构变成分子关系网,精准捕捉远程相互作用
终极愿景:当这些技术熔铸一炉,ProMEP将成为生物版的「ChatGPT」——输入目标功能,输出最优突变方案。从癌症治疗到碳中和酶设计,人类终于握住了改写生命蓝图的「代码钢笔」! ✍️🔬
赛博修仙版(科研放松时刻):