转录组数据挖掘(生物技能树)(第11节)
R语言复习

#### 读取 ####
dat = read.delim("GSE188603_genes_fpkm_expression.txt.gz",header = F)
#发现文件不规范,需要练习整理数据
dat[1,]
#因为不正常,才加处理的步骤
### 跳过第一行来读取
dat = read.delim("GSE188603_genes_fpkm_expression.txt.gz",skip = 1,header = F) #skip = 1 跳过第一行,header = F 列名不设置
##获取文件第一行的内容
f = readLines("GSE188603_genes_fpkm_expression.txt.gz") #获取字符串
f[1] #查看f的第一行
library(stringr)
cn = str_split(f[1],"\t| ")[[1]] # str_split 实现字符串的拆分
cn = cn[cn!=""] #不要那种空字符串的,把列名拆分出来
cn
colnames(dat) = cn
#因为不正常,才加处理的步骤
## 第二种读取文件的方法
dat2 = data.table::fread("GSE188603_genes_fpkm_expression.txt.gz",data.table = F)
colnames(dat2) = cn
## 第三种读取文件的方法
dat3 = rio::import("GSE188603_genes_fpkm_expression.txt.gz")
colnames(dat3) = cn
#### 数据框 ####
#取子集
mdat = dat[1:10,]
mdat$gene_id
mdat[,"gene_id"]
mdat[1:2,3:4]
#转换成矩阵
#去除不是数值的列
#一个规律,当一个数据里同时出现ensemlel id和symbol时,通常ensemlel id没有重复,symbol有重复
#验证
length(unique(dat$gene_id)) #看看跟行数是不是一样多
table(duplicated(dat$gene_id))
#row.names = 1
#怎么排查有什么异常
head(sort(table(dat$gene_name),decreasing = T))
Y_RNA Metazoa_SRP U3 U6 SNORA70 U2
622 142 42 26 25 19
#去掉前两个,看起来不正常
k1 = dat$gene_name == c("Y_RNA","Metazoa_SRP");table(k1) #用==不对,加起来的总和和上边的对不上
k = dat$gene_name %in% c("Y_RNA","Metazoa_SRP");table(k) #用%in% 就全部选上
dat = dat[!k,] #取子集,去掉一些
#想把gene_name设为行名,要先去重复
library(dplyr)
dat = distinct(dat, gene_name, .keep_all = T) #使用distinct()函数去除数据框dat中gene_name列的重复行,同时保留所有其他列。
exp = as.matrix(dat[,-c(1,2)])
rownames(exp) = dat$gene_name #把数据框转换成矩阵
### 矩阵 ###
### 如何把表达量为0的gene去掉(行和为0)
#apply(exp,1, sum)
k2 = rowSums(exp) > 0;table(k2) #计算每一行的总和,选出大于0的行
exp = exp[k2,]
#方差最大的500个基因
gs = names(tail(sort(apply(exp, 1, var)),50)) #var取方差
n = exp[gs,]
pheatmap::pheatmap(n) #画图后发现太丑是由于没取log
n= 1og2(exp[gs,]+1) #再取log
pheatmap::pheatmap(n)#重新画图,结果图还是丑
exp = 1og2(exp+1) #再次尝试先取log,再求方差
gs = names(tail(sort(apply(exp, 1, var)),50))
n = exp[gs,]
pheatmap::pheatmap(n)#重新画图,正常
pheatmap::pheatmap(n,
scale ="row") #加个参数,使得颜色更鲜明,按行进行标准化
library(tinyarray)
colnames(n) #查看分组信息
Group = c("HCQ","HCQ","NG","NG")
Group = factor(Group, levels = c("HCQ","NG"))
draw_heatmap(n,Group)
draw_pca(n,Group)
draw_boxplot(n[1:10,],Group)
wilcox.test(n[1,]~Group)
draw_tsne(n,Group)

转录组数据差异分析
** limma **
** edgeR **
** DESeq2 **
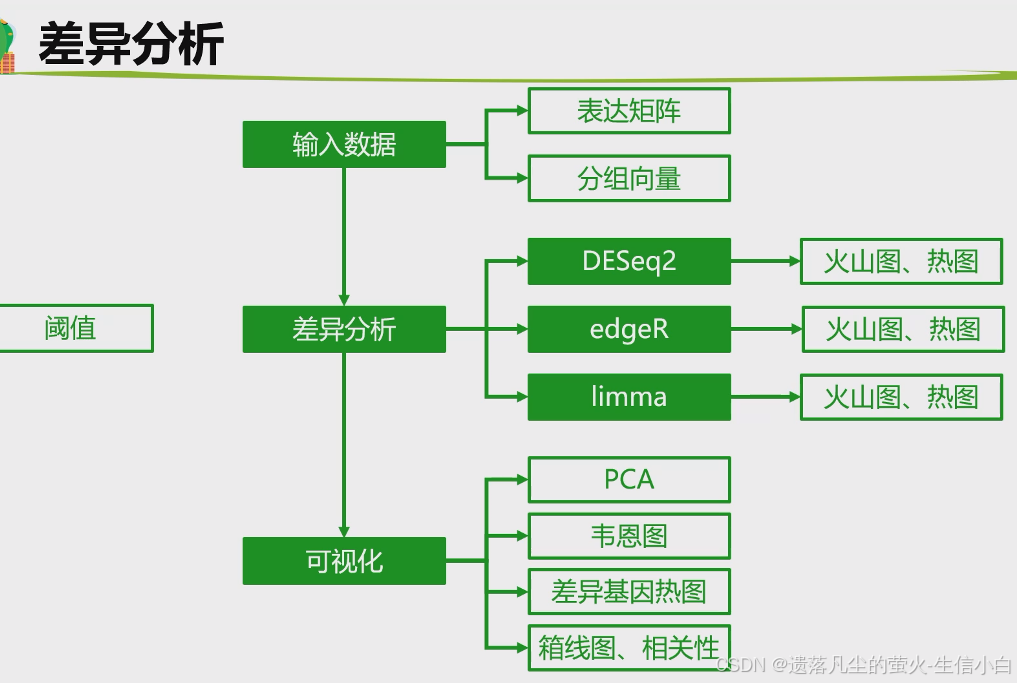

差异分析的输入数据
** 表达矩阵 **
• 数值型矩阵-count
• 行名是symbol
• 低表达量的基因要过滤掉
** 分组信息 **
• 因子,对照组在level第一位
• 与表达矩阵的列一一对应
对于GEO来说,分组信息藏在临床信息表格里,对于其他数据库,分组信息可能藏在列名里
** 项目名称 **
• 字符串
• TCGA-XXXX
• 非TCGA数据特殊无要求
芯片数据的输入数据: 表达矩阵,分组信息,探针注释

操作过程
打开case1 pipeline文件夹, 双击里面的rproj文件,从右下角打开1 data.Rmd
安装过时的包:
install.packages("https://cran.r-project.org/src/contrib/Archive/deconstructSigs/deconstructSigs_1.8.0.tar.gz",
repos = NULL,dependencies = T)
示例一:
来自UCSC Xena的一个数据,不能算是TCGA的标准做法
1.data
生信技能树
1.起个项目名字
TCGA的数据,统一叫TCGA-xxxx,非TCGA的数据随意起名,不要有特殊字符即可。
proj = "TCGA-CHOL"
2.读取和整理数据
2.1 表达矩阵
dat = read.table("TCGA-CHOL.htseq_counts.tsv.gz",check.names = F,row.names = 1,header = T)
range(dat)
#> [1] 0.0000 24.1811
#逆转log,发现需要逆转,才逆转
dat = as.matrix(2^dat - 1)
dat[1:4,1:4]
#> TCGA-ZD-A8I3-01A TCGA-W5-AA2U-11A TCGA-W5-AA30-01A
#> ENSG00000000003.13 5254 2476 5132
#> ENSG00000000005.5 1 1 0
#> ENSG00000000419.11 1212 655 1644
#> ENSG00000000457.12 753 346 2652
#> TCGA-W5-AA38-01A
#> ENSG00000000003.13 8249
#> ENSG00000000005.5 1
#> ENSG00000000419.11 1696
#> ENSG00000000457.12 519
# 深坑一个
dat[97,9]
#> [1] 876
as.character(dat[97,9]) #眼见不一定为实吧。
#> [1] "875.999999999999"
# 转换为整数矩阵
exp = round(dat)
# 检查
as.character(exp[97,9])
#> [1] "876"
2.2 临床信息
clinical = read.delim("TCGA-CHOL.GDC_phenotype.tsv.gz")
clinical[1:4,1:4]
#> submitter_id.samples age_at_initial_pathologic_diagnosis
#> 1 TCGA-ZH-A8Y2-01A 59
#> 2 TCGA-ZH-A8Y7-01A 59
#> 3 TCGA-W7-A93O-01A NA
#> 4 TCGA-W7-A93O-11A NA
#> albumin_result_lower_limit albumin_result_specified_value
#> 1 NA NA
#> 2 3.5 2.4
#> 3 NA NA
#> 4 NA NA
3.表达矩阵行名ID转换
library(tinyarray)
#> Warning: package 'tinyarray' was built under R version 4.3.3
exp = trans_exp_new(exp)
#> Warning in AnnoProbe::annoGene(rownames(exp), ID_type = "ENSEMBL", species =
#> species): 6.54% of input IDs are fail to annotate...
exp[1:4,1:4]
#> TCGA-ZD-A8I3-01A TCGA-W5-AA2U-11A TCGA-W5-AA30-01A TCGA-W5-AA38-01A
#> DDX11L1 0 0 0 1
#> WASH7P 81 10 146 55
#> MIR6859-1 1 0 11 1
#> MIR1302-2HG 0 0 0 0
4.基因过滤
需要过滤一下那些在很多样本里表达量都为0或者表达量很低的基因。过滤标准不唯一。
过滤之前基因数量:
nrow(exp)
#> [1] 56514
常用过滤标准1:
仅去除在所有样本里表达量都为零的基因
exp1 = exp[rowSums(exp)>0,]
nrow(exp1)
#> [1] 48057
常用过滤标准2(推荐):
仅保留在一半以上样本里表达的基因
exp = exp[apply(exp, 1, function(x) sum(x > 0) > 0.5*ncol(exp)), ]
nrow(exp)
#> [1] 28434
5.分组信息获取
TCGA的数据,直接用make_tcga_group给样本分组(tumor和normal),其他地方的数据分组方式参考芯片数据pipeline/02_group_ids.R
library(tinyarray)
Group = make_tcga_group(exp)
table(Group)
#> Group
#> normal tumor
#> 9 36
6.保存数据
save(exp,Group,proj,clinical,file = paste0(proj,".Rdata"))
三大R包差异分析
1.三大R包差异分析
rm(list = ls())
load("TCGA-CHOL.Rdata")
table(Group)
#> Group
#> normal tumor
#> 9 36
#deseq2----
library(DESeq2)
colData <- data.frame(row.names =colnames(exp),
condition=Group)
#注意事项:如果多次运行,表达矩阵改动过的话,需要从工作目录下删除下面if括号里的文件
if(!file.exists(paste0(proj,"_dd.Rdata"))){
dds <- DESeqDataSetFromMatrix(
countData = exp,
colData = colData,
design = ~ condition)
dds <- DESeq(dds)
save(dds,file = paste0(proj,"_dd.Rdata"))
}
load(file = paste0(proj,"_dd.Rdata"))
class(dds)
#> [1] "DESeqDataSet"
#> attr(,"package")
#> [1] "DESeq2"
res <- results(dds, contrast = c("condition",rev(levels(Group))))
#constrast
c("condition",rev(levels(Group)))
#> [1] "condition" "tumor" "normal"
class(res)
#> [1] "DESeqResults"
#> attr(,"package")
#> [1] "DESeq2"
DEG1 <- as.data.frame(res)
library(dplyr)
DEG1 <- arrange(DEG1,pvalue)
DEG1 = na.omit(DEG1)
head(DEG1)
#> baseMean log2FoldChange lfcSE stat pvalue padj
#> GCDH 2294.6422 -3.311000 0.1837627 -18.01781 1.412303e-72 4.015744e-68
#> MSMO1 7536.4635 -3.769054 0.2187601 -17.22916 1.604644e-66 2.281322e-62
#> KCNN2 468.3807 -5.607502 0.3344614 -16.76577 4.343673e-63 3.433737e-59
#> TCAIM 1255.8164 -2.232825 0.1332278 -16.75945 4.830467e-63 3.433737e-59
#> USH2A 549.5469 -6.473181 0.3879584 -16.68524 1.678162e-62 9.543371e-59
#> RCL1 1642.8797 -3.842743 0.2345231 -16.38535 2.433482e-60 1.153227e-56
#添加change列标记基因上调下调
logFC_t = 2
pvalue_t = 0.05
k1 = (DEG1$pvalue < pvalue_t)&(DEG1$log2FoldChange < -logFC_t);table(k1)
#> k1
#> FALSE TRUE
#> 26378 2056
k2 = (DEG1$pvalue < pvalue_t)&(DEG1$log2FoldChange > logFC_t);table(k2)
#> k2
#> FALSE TRUE
#> 24590 3844
DEG1$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))
table(DEG1$change)
#>
#> DOWN NOT UP
#> 2056 22534 3844
head(DEG1)
#> baseMean log2FoldChange lfcSE stat pvalue padj
#> GCDH 2294.6422 -3.311000 0.1837627 -18.01781 1.412303e-72 4.015744e-68
#> MSMO1 7536.4635 -3.769054 0.2187601 -17.22916 1.604644e-66 2.281322e-62
#> KCNN2 468.3807 -5.607502 0.3344614 -16.76577 4.343673e-63 3.433737e-59
#> TCAIM 1255.8164 -2.232825 0.1332278 -16.75945 4.830467e-63 3.433737e-59
#> USH2A 549.5469 -6.473181 0.3879584 -16.68524 1.678162e-62 9.543371e-59
#> RCL1 1642.8797 -3.842743 0.2345231 -16.38535 2.433482e-60 1.153227e-56
#> change
#> GCDH DOWN
#> MSMO1 DOWN
#> KCNN2 DOWN
#> TCAIM DOWN
#> USH2A DOWN
#> RCL1 DOWN
#edgeR----
library(edgeR)
dge <- DGEList(counts=exp,group=Group)
dge$samples$lib.size <- colSums(dge$counts)
dge <- calcNormFactors(dge)
design <- model.matrix(~Group)
dge <- estimateGLMCommonDisp(dge, design)
dge <- estimateGLMTrendedDisp(dge, design)
dge <- estimateGLMTagwiseDisp(dge, design)
fit <- glmFit(dge, design)
fit <- glmLRT(fit)
DEG2=topTags(fit, n=Inf)
class(DEG2)
#> [1] "TopTags"
#> attr(,"package")
#> [1] "edgeR"
DEG2=as.data.frame(DEG2)
head(DEG2)
#> logFC logCPM LR PValue FDR
#> USH2A -6.378447 3.538793 347.2383 1.692535e-77 4.812553e-73
#> KCNN2 -5.491333 3.311899 318.4008 3.230333e-71 4.592565e-67
#> GCDH -3.209015 5.626247 312.2237 7.158377e-70 6.784710e-66
#> SC5D -4.080367 6.277540 305.5612 2.023988e-68 1.438752e-64
#> LCAT -4.650620 6.387429 304.4307 3.568629e-68 2.029408e-64
#> RCL1 -3.739003 5.135921 302.7227 8.406025e-68 3.983615e-64
k1 = (DEG2$PValue < pvalue_t)&(DEG2$logFC < -logFC_t)
k2 = (DEG2$PValue < pvalue_t)&(DEG2$logFC > logFC_t)
DEG2$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))
head(DEG2)
#> logFC logCPM LR PValue FDR change
#> USH2A -6.378447 3.538793 347.2383 1.692535e-77 4.812553e-73 DOWN
#> KCNN2 -5.491333 3.311899 318.4008 3.230333e-71 4.592565e-67 DOWN
#> GCDH -3.209015 5.626247 312.2237 7.158377e-70 6.784710e-66 DOWN
#> SC5D -4.080367 6.277540 305.5612 2.023988e-68 1.438752e-64 DOWN
#> LCAT -4.650620 6.387429 304.4307 3.568629e-68 2.029408e-64 DOWN
#> RCL1 -3.739003 5.135921 302.7227 8.406025e-68 3.983615e-64 DOWN
table(DEG2$change)
#>
#> DOWN NOT UP
#> 1870 22030 4534
#limma----
library(limma)
dge <- edgeR::DGEList(counts=exp)
dge <- edgeR::calcNormFactors(dge)
design <- model.matrix(~Group)
v <- voom(dge,design, normalize="quantile")
fit <- lmFit(v, design)
fit= eBayes(fit)
DEG3 = topTable(fit, coef=2, n=Inf)
DEG3 = na.omit(DEG3)
k1 = (DEG3$P.Value < pvalue_t)&(DEG3$logFC < -logFC_t)
k2 = (DEG3$P.Value < pvalue_t)&(DEG3$logFC > logFC_t)
DEG3$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))
table(DEG3$change)
#>
#> DOWN NOT UP
#> 2418 24089 1927
head(DEG3)
#> logFC AveExpr t P.Value adj.P.Val B change
#> USH2A -6.465514 0.2876576 -23.44588 1.005120e-27 2.857959e-23 52.37631 DOWN
#> KCNN2 -5.525147 0.7856401 -21.43855 4.886457e-26 6.947076e-22 48.72067 DOWN
#> FXYD1 -6.885491 0.5960549 -20.80430 1.774229e-25 1.681614e-21 47.55727 DOWN
#> ASPDH -7.429138 1.0233269 -19.13958 6.120565e-24 4.350804e-20 44.20531 DOWN
#> ESR1 -5.690485 1.2749510 -18.62076 1.938371e-23 1.012411e-19 43.05941 DOWN
#> RMDN2 -2.832609 2.4413375 -18.53491 2.351336e-23 1.012411e-19 42.85041 DOWN
tj = data.frame(deseq2 = as.integer(table(DEG1$change)),
edgeR = as.integer(table(DEG2$change)),
limma_voom = as.integer(table(DEG3$change)),
row.names = c("down","not","up")
);tj
#> deseq2 edgeR limma_voom
#> down 2056 1870 2418
#> not 22534 22030 24089
#> up 3844 4534 1927
save(DEG1,DEG2,DEG3,Group,tj,file = paste0(proj,"_DEG.Rdata"))
2.可视化
library(ggplot2)
library(tinyarray)
exp[1:4,1:4]
#> TCGA-ZD-A8I3-01A TCGA-W5-AA2U-11A TCGA-W5-AA30-01A TCGA-W5-AA38-01A
#> WASH7P 81 10 146 55
#> MIR6859-1 1 0 11 1
#> CICP27 0 1 2 1
#> AL627309.6 37 4 45 41
# cpm 去除文库大小的影响
dat = log2(cpm(exp)+1)
pca.plot = draw_pca(dat,Group);pca.plot
save(pca.plot,file = paste0(proj,"_pcaplot.Rdata"))
cg1 = rownames(DEG1)[DEG1$change !="NOT"]
cg2 = rownames(DEG2)[DEG2$change !="NOT"]
cg3 = rownames(DEG3)[DEG3$change !="NOT"]
h1 = draw_heatmap(dat[cg1,],Group)
h2 = draw_heatmap(dat[cg2,],Group)
h3 = draw_heatmap(dat[cg3,],Group)
v1 = draw_volcano(DEG1,pkg = 1,logFC_cutoff = logFC_t)
v2 = draw_volcano(DEG2,pkg = 2,logFC_cutoff = logFC_t)
v3 = draw_volcano(DEG3,pkg = 3,logFC_cutoff = logFC_t)
library(patchwork)
(h1 + h2 + h3) / (v1 + v2 + v3) +plot_layout(guides = 'collect') &theme(legend.position = "none")
ggsave(paste0(proj,"_heat_vo.png"),width = 15,height = 10)
3.三大R包差异基因对比
UP=function(df){
rownames(df)[df$change=="UP"]
}
DOWN=function(df){
rownames(df)[df$change=="DOWN"]
}
up = intersect(intersect(UP(DEG1),UP(DEG2)),UP(DEG3))
down = intersect(intersect(DOWN(DEG1),DOWN(DEG2)),DOWN(DEG3))
dat = log2(cpm(exp)+1)
hp = draw_heatmap(dat[c(up,down),],Group)
#上调、下调基因分别画维恩图
up_genes = list(Deseq2 = UP(DEG1),
edgeR = UP(DEG2),
limma = UP(DEG3))
down_genes = list(Deseq2 = DOWN(DEG1),
edgeR = DOWN(DEG2),
limma = DOWN(DEG3))
up.plot <- draw_venn(up_genes,"UPgene")
down.plot <- draw_venn(down_genes,"DOWNgene")
#维恩图拼图,终于搞定
library(patchwork)
#up.plot + down.plot
# 拼图
pca.plot + hp+up.plot +down.plot+ plot_layout(guides = "collect")
ggsave(paste0(proj,"_heat_ve_pca.png"),width = 15,height = 10)
分组聚类的热图
library(ComplexHeatmap)
library(circlize)
col_fun = colorRamp2(c(-2, 0, 2), c("#2fa1dd", "white", "#f87669"))
top_annotation = HeatmapAnnotation(
cluster = anno_block(gp = gpar(fill = c("#f87669","#2fa1dd")),
labels = levels(Group),
labels_gp = gpar(col = "white", fontsize = 12)))
m = Heatmap(t(scale(t(exp[c(up,down),]))),name = " ",
col = col_fun,
top_annotation = top_annotation,
column_split = Group,
show_heatmap_legend = T,
border = F,
show_column_names = F,
show_row_names = F,
use_raster = F,
cluster_column_slices = F,
column_title = NULL)
m
示例二:
打开Day11-5.20-case_2_GSE193861-geo_rnaseq
1.data
生信技能树
1.起个项目名字
TCGA的数据,统一叫TCGA-xxxx,非TCGA的数据随意起名,不要有特殊字符即可。
proj = "GSE193861"
2.读取和整理数据
2.1 表达矩阵
# 读取一个文件
b = "GSM5822748_con1.txt.gz"
# 列名后加
r1 = function(b){
read.delim(paste0("GSE193861_RAW/",b),header = F,row.names = 1)
}
bs = dir("GSE193861_RAW/")
dat = lapply(bs, r1)
#新函数 do.call 对列表进行批量操作
exp = do.call(cbind,dat)
#列名,从bs的文件名称里提取,\\.代表指.号本身,而不是正则表达式
library(stringr)
#> Warning: package 'stringr' was built under R version 4.3.3
colnames(exp) = str_split_i(bs,"_|\\.",2)
2.2 临床信息
library(tinyarray) clinical = geo_download(“GSE193861”, colon_remove = T)$pd,也可以加上colon_remove = T去掉多余的列
library(tinyarray)
#> Warning: package 'tinyarray' was built under R version 4.3.3
clinical = geo_download("GSE193861")$pd
#> Warning in geo_download("GSE193861"): exp is empty
3.表达矩阵行名ID转换
library(tinyarray)
exp = trans_exp_new(exp)
#> Warning in AnnoProbe::annoGene(rownames(exp), ID_type = "ENSEMBL", species =
#> species): 17.79% of input IDs are fail to annotate...
exp[1:4,1:4]
#> con1 con2 con3 con4
#> DDX11L1 0 0 0 0
#> WASH7P 30 11 4 33
#> MIR1302-2HG 0 0 0 0
#> FAM138A 0 0 0 0
4.基因过滤
需要过滤一下那些在很多样本里表达量都为0或者表达量很低的基因。过滤标准不唯一。
过滤之前基因数量:
nrow(exp)
#> [1] 52343
常用过滤标准1:
仅去除在所有样本里表达量都为零的基因
exp1 = exp[rowSums(exp)>0,]
nrow(exp1)
#> [1] 31424
常用过滤标准2(推荐):
仅保留在一半以上样本里表达的基因
exp = exp[apply(exp, 1, function(x) sum(x > 0) > 0.5*ncol(exp)), ]
nrow(exp)
#> [1] 20383
5.分组信息获取
TCGA的数据,直接用make_tcga_group给样本分组(tumor和normal),其他地方的数据分组方式参考芯片数据pipeline/02_group_ids.R
colnames(exp)
#> [1] "con1" "con2" "con3" "con4" "con5" "DOX1" "DOX2" "DOX3" "DOX4" "DOX6"
library(stringr)
Group = str_remove_all(colnames(exp),"\\d")
Group = factor(Group,levels = c("con","DOX"))
table(Group)
#> Group
#> con DOX
#> 5 5
6.保存数据
save(exp,Group,proj,clinical,file = paste0(proj,".Rdata"))
三大R包差异分析
1.三大R包差异分析
rm(list = ls())
load("GSE193861.Rdata")
table(Group)
#> Group
#> con DOX
#> 5 5
#deseq2----
library(DESeq2)
colData <- data.frame(row.names =colnames(exp),
condition=Group)
#注意事项:如果多次运行,表达矩阵改动过的话,需要从工作目录下删除下面if括号里的文件
if(!file.exists(paste0(proj,"_dd.Rdata"))){
dds <- DESeqDataSetFromMatrix(
countData = exp,
colData = colData,
design = ~ condition)
dds <- DESeq(dds)
save(dds,file = paste0(proj,"_dd.Rdata"))
}
load(file = paste0(proj,"_dd.Rdata"))
class(dds)
#> [1] "DESeqDataSet"
#> attr(,"package")
#> [1] "DESeq2"
res <- results(dds, contrast = c("condition",rev(levels(Group))))
#constrast
c("condition",rev(levels(Group)))
#> [1] "condition" "DOX" "con"
class(res)
#> [1] "DESeqResults"
#> attr(,"package")
#> [1] "DESeq2"
DEG1 <- as.data.frame(res)
library(dplyr)
DEG1 <- arrange(DEG1,pvalue)
DEG1 = na.omit(DEG1)
head(DEG1)
#> baseMean log2FoldChange lfcSE stat pvalue
#> XIST 1113.44045 7.745398 1.1590688 6.682431 2.350101e-11
#> RELN 73.77183 3.181230 0.5878329 5.411793 6.239663e-08
#> MYH6 5722.85394 -2.937321 0.5486449 -5.353774 8.613828e-08
#> MME 89.49695 2.608529 0.5694700 4.580625 4.635893e-06
#> MTCO2P12 54.33513 4.692185 1.0293695 4.558310 5.156698e-06
#> SNORA65 40.60296 4.697523 1.0357679 4.535305 5.752034e-06
#> padj
#> XIST 3.659577e-07
#> RELN 4.471151e-04
#> MYH6 4.471151e-04
#> MME 1.492844e-02
#> MTCO2P12 1.492844e-02
#> SNORA65 1.492844e-02
#添加change列标记基因上调下调
logFC_t = 1
pvalue_t = 0.05
k1 = (DEG1$pvalue < pvalue_t)&(DEG1$log2FoldChange < -logFC_t);table(k1)
#> k1
#> FALSE TRUE
#> 15466 106
k2 = (DEG1$pvalue < pvalue_t)&(DEG1$log2FoldChange > logFC_t);table(k2)
#> k2
#> FALSE TRUE
#> 15268 304
DEG1$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))
table(DEG1$change)
#>
#> DOWN NOT UP
#> 106 15162 304
head(DEG1)
#> baseMean log2FoldChange lfcSE stat pvalue
#> XIST 1113.44045 7.745398 1.1590688 6.682431 2.350101e-11
#> RELN 73.77183 3.181230 0.5878329 5.411793 6.239663e-08
#> MYH6 5722.85394 -2.937321 0.5486449 -5.353774 8.613828e-08
#> MME 89.49695 2.608529 0.5694700 4.580625 4.635893e-06
#> MTCO2P12 54.33513 4.692185 1.0293695 4.558310 5.156698e-06
#> SNORA65 40.60296 4.697523 1.0357679 4.535305 5.752034e-06
#> padj change
#> XIST 3.659577e-07 UP
#> RELN 4.471151e-04 UP
#> MYH6 4.471151e-04 DOWN
#> MME 1.492844e-02 UP
#> MTCO2P12 1.492844e-02 UP
#> SNORA65 1.492844e-02 UP
#edgeR----
library(edgeR)
dge <- DGEList(counts=exp,group=Group)
dge$samples$lib.size <- colSums(dge$counts)
dge <- calcNormFactors(dge)
design <- model.matrix(~Group)
dge <- estimateGLMCommonDisp(dge, design)
dge <- estimateGLMTrendedDisp(dge, design)
dge <- estimateGLMTagwiseDisp(dge, design)
fit <- glmFit(dge, design)
fit <- glmLRT(fit)
DEG2=topTags(fit, n=Inf)
class(DEG2)
#> [1] "TopTags"
#> attr(,"package")
#> [1] "edgeR"
DEG2=as.data.frame(DEG2)
head(DEG2)
#> logFC logCPM LR PValue FDR
#> XIST 7.726373 6.1664739 40.20227 2.289819e-10 4.667337e-06
#> RELN 3.142138 2.1513189 35.75147 2.241633e-09 2.284560e-05
#> ADIPOQ 6.768696 1.9737844 31.43692 2.060262e-08 1.399811e-04
#> UNC80 5.578451 -0.3805617 30.80077 2.859249e-08 1.457002e-04
#> PLPPR4 4.152559 -1.2836374 29.23704 6.404322e-08 2.610786e-04
#> MMRN1 3.066753 0.8954184 28.74065 8.274826e-08 2.811096e-04
k1 = (DEG2$PValue < pvalue_t)&(DEG2$logFC < -logFC_t)
k2 = (DEG2$PValue < pvalue_t)&(DEG2$logFC > logFC_t)
DEG2$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))
head(DEG2)
#> logFC logCPM LR PValue FDR change
#> XIST 7.726373 6.1664739 40.20227 2.289819e-10 4.667337e-06 UP
#> RELN 3.142138 2.1513189 35.75147 2.241633e-09 2.284560e-05 UP
#> ADIPOQ 6.768696 1.9737844 31.43692 2.060262e-08 1.399811e-04 UP
#> UNC80 5.578451 -0.3805617 30.80077 2.859249e-08 1.457002e-04 UP
#> PLPPR4 4.152559 -1.2836374 29.23704 6.404322e-08 2.610786e-04 UP
#> MMRN1 3.066753 0.8954184 28.74065 8.274826e-08 2.811096e-04 UP
table(DEG2$change)
#>
#> DOWN NOT UP
#> 344 19256 783
#limma----
library(limma)
dge <- edgeR::DGEList(counts=exp)
dge <- edgeR::calcNormFactors(dge)
design <- model.matrix(~Group)
v <- voom(dge,design, normalize="quantile")
fit <- lmFit(v, design)
fit= eBayes(fit)
DEG3 = topTable(fit, coef=2, n=Inf)
DEG3 = na.omit(DEG3)
k1 = (DEG3$P.Value < pvalue_t)&(DEG3$logFC < -logFC_t)
k2 = (DEG3$P.Value < pvalue_t)&(DEG3$logFC > logFC_t)
DEG3$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))
table(DEG3$change)
#>
#> DOWN NOT UP
#> 200 19743 440
head(DEG3)
#> logFC AveExpr t P.Value adj.P.Val B change
#> RYR2 -1.602209 10.186360 -4.087521 0.0013467451 0.9820625 -3.140559 DOWN
#> MT-ND5 1.437690 10.392968 3.664636 0.0029671911 0.9820625 -3.263672 UP
#> MT-ND3 1.191078 9.677601 2.933701 0.0118911929 0.9940732 -3.709765 UP
#> DES -1.004170 10.901441 -2.588390 0.0228599216 0.9940732 -3.797395 DOWN
#> XIST 6.385181 2.303796 5.287750 0.0001594876 0.5418061 -3.840880 UP
#> PDK4 2.835272 6.800228 3.363489 0.0052476916 0.9940732 -3.891868 UP
tj = data.frame(deseq2 = as.integer(table(DEG1$change)),
edgeR = as.integer(table(DEG2$change)),
limma_voom = as.integer(table(DEG3$change)),
row.names = c("down","not","up")
);tj
#> deseq2 edgeR limma_voom
#> down 106 344 200
#> not 15162 19256 19743
#> up 304 783 440
save(DEG1,DEG2,DEG3,Group,tj,file = paste0(proj,"_DEG.Rdata"))
** 2.可视化 **
library(ggplot2)
library(tinyarray)
exp[1:4,1:4]
#> con1 con2 con3 con4
#> WASH7P 30 11 4 33
#> AP006222.1 6 2 2 1
#> MTND1P23 2 4 4 6
#> MTND2P28 2 1704 734 1001
# cpm 去除文库大小的影响
dat = log2(cpm(exp)+1)
pca.plot = draw_pca(dat,Group);pca.plot
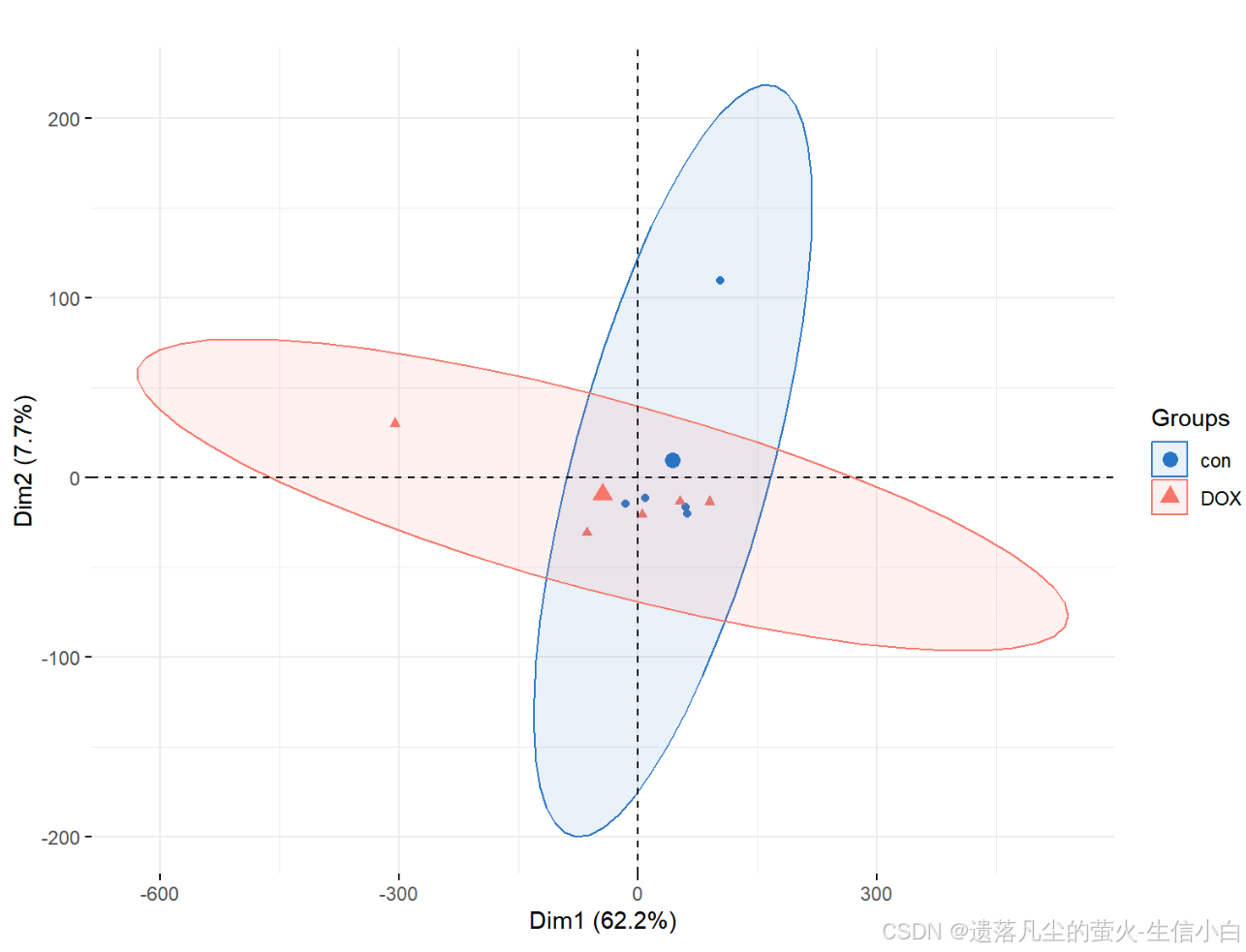
save(pca.plot,file = paste0(proj,"_pcaplot.Rdata"))
cg1 = rownames(DEG1)[DEG1$change !="NOT"]
cg2 = rownames(DEG2)[DEG2$change !="NOT"]
cg3 = rownames(DEG3)[DEG3$change !="NOT"]
h1 = draw_heatmap(dat[cg1,],Group)
h2 = draw_heatmap(dat[cg2,],Group)
h3 = draw_heatmap(dat[cg3,],Group)
v1 = draw_volcano(DEG1,pkg = 1,logFC_cutoff = logFC_t)
v2 = draw_volcano(DEG2,pkg = 2,logFC_cutoff = logFC_t)
v3 = draw_volcano(DEG3,pkg = 3,logFC_cutoff = logFC_t)
library(patchwork)
(h1 + h2 + h3) / (v1 + v2 + v3) +plot_layout(guides = 'collect') &theme(legend.position = "none")
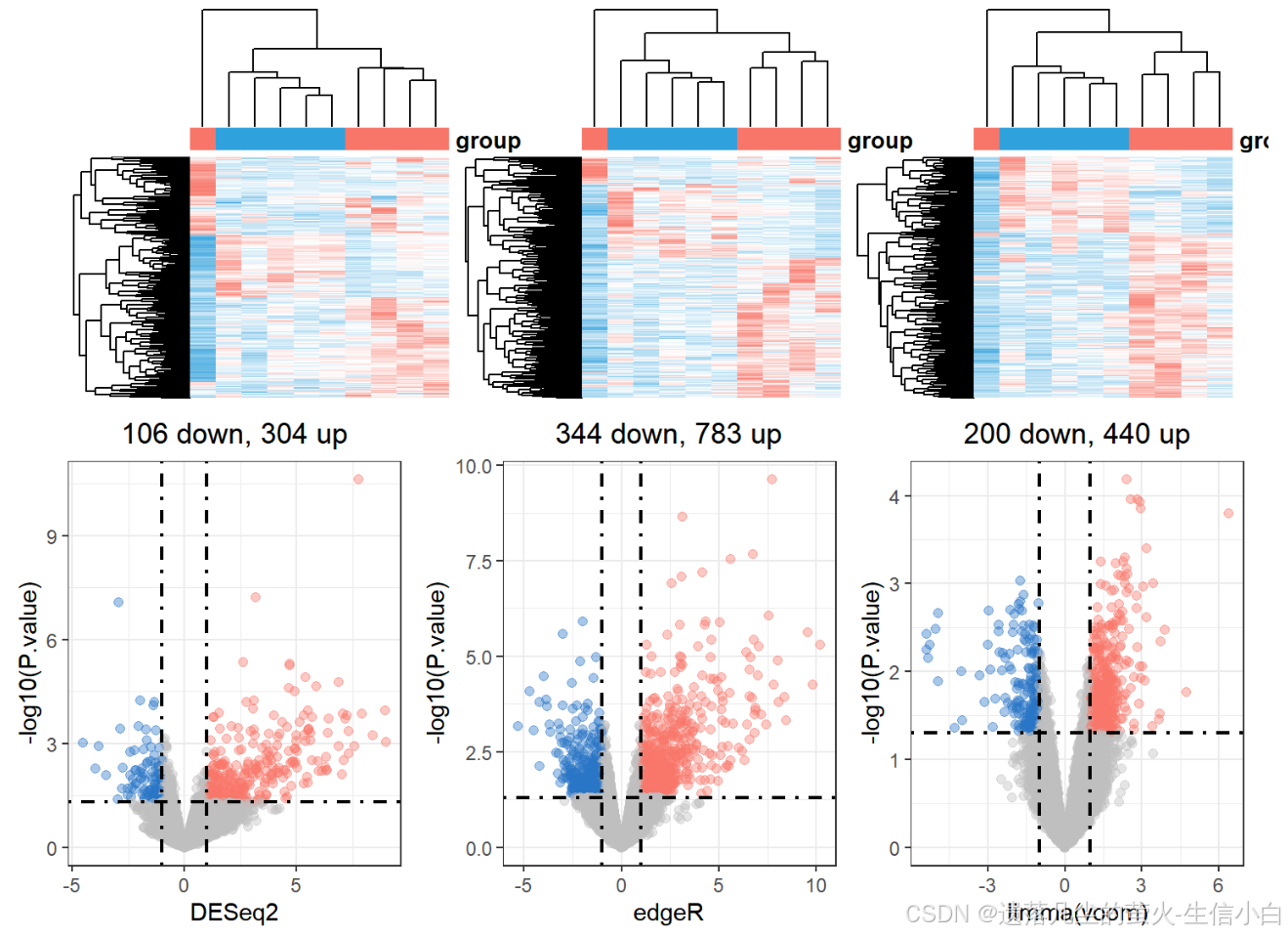
ggsave(paste0(proj,"_heat_vo.png"),width = 15,height = 10)
3.三大R包差异基因对比
UP=function(df){
rownames(df)[df$change=="UP"]
}
DOWN=function(df){
rownames(df)[df$change=="DOWN"]
}
up = intersect(intersect(UP(DEG1),UP(DEG2)),UP(DEG3))
down = intersect(intersect(DOWN(DEG1),DOWN(DEG2)),DOWN(DEG3))
dat = log2(cpm(exp)+1)
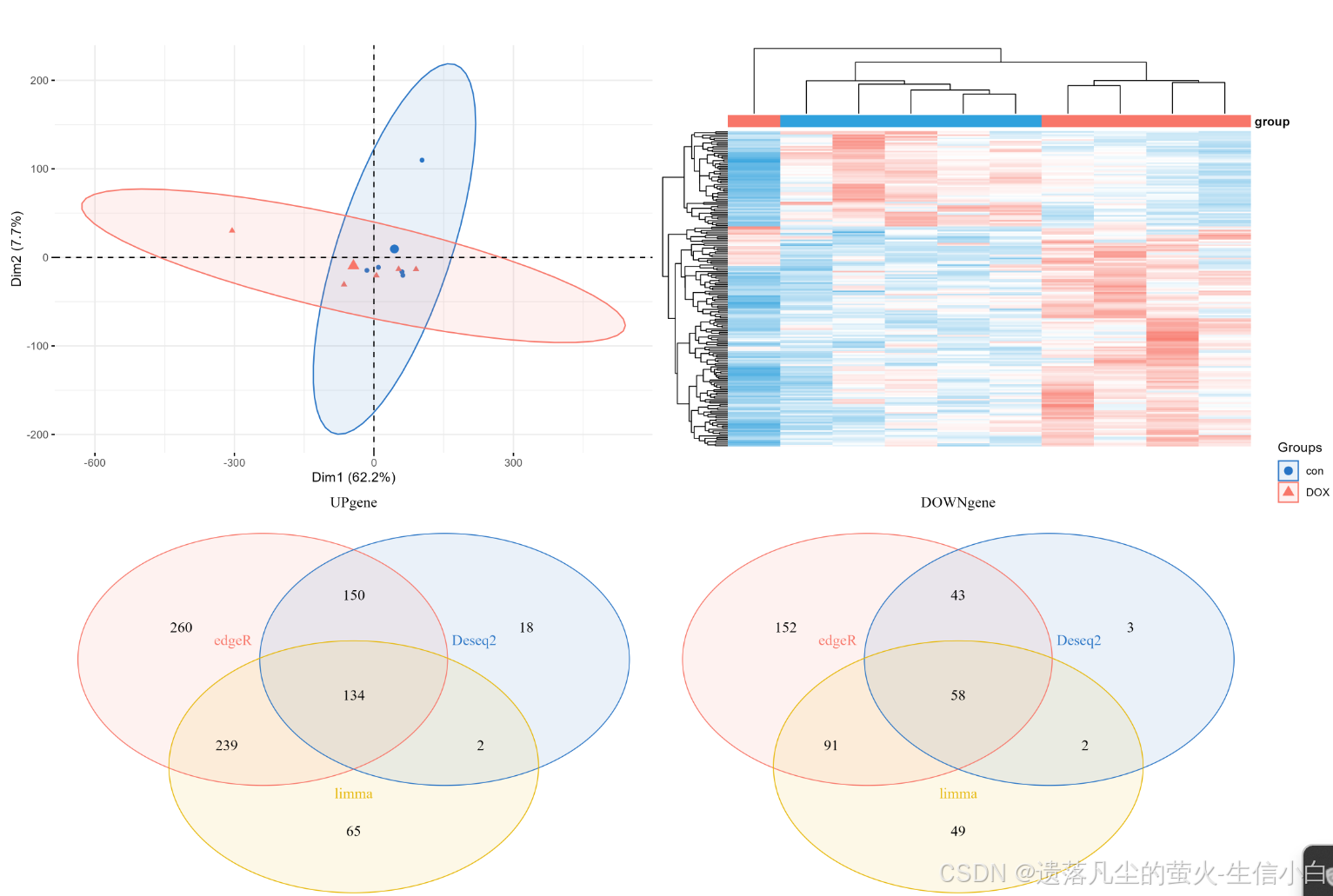
hp = draw_heatmap(dat[c(up,down),],Group)
#上调、下调基因分别画维恩图
up_genes = list(Deseq2 = UP(DEG1),
edgeR = UP(DEG2),
limma = UP(DEG3))
down_genes = list(Deseq2 = DOWN(DEG1),
edgeR = DOWN(DEG2),
limma = DOWN(DEG3))
up.plot <- draw_venn(up_genes,"UPgene")
down.plot <- draw_venn(down_genes,"DOWNgene")
#维恩图拼图,终于搞定
library(patchwork)
#up.plot + down.plot
# 拼图
pca.plot + hp+up.plot +down.plot+ plot_layout(guides = "collect")
ggsave(paste0(proj,"_heat_ve_pca.png"),width = 15,height = 10)
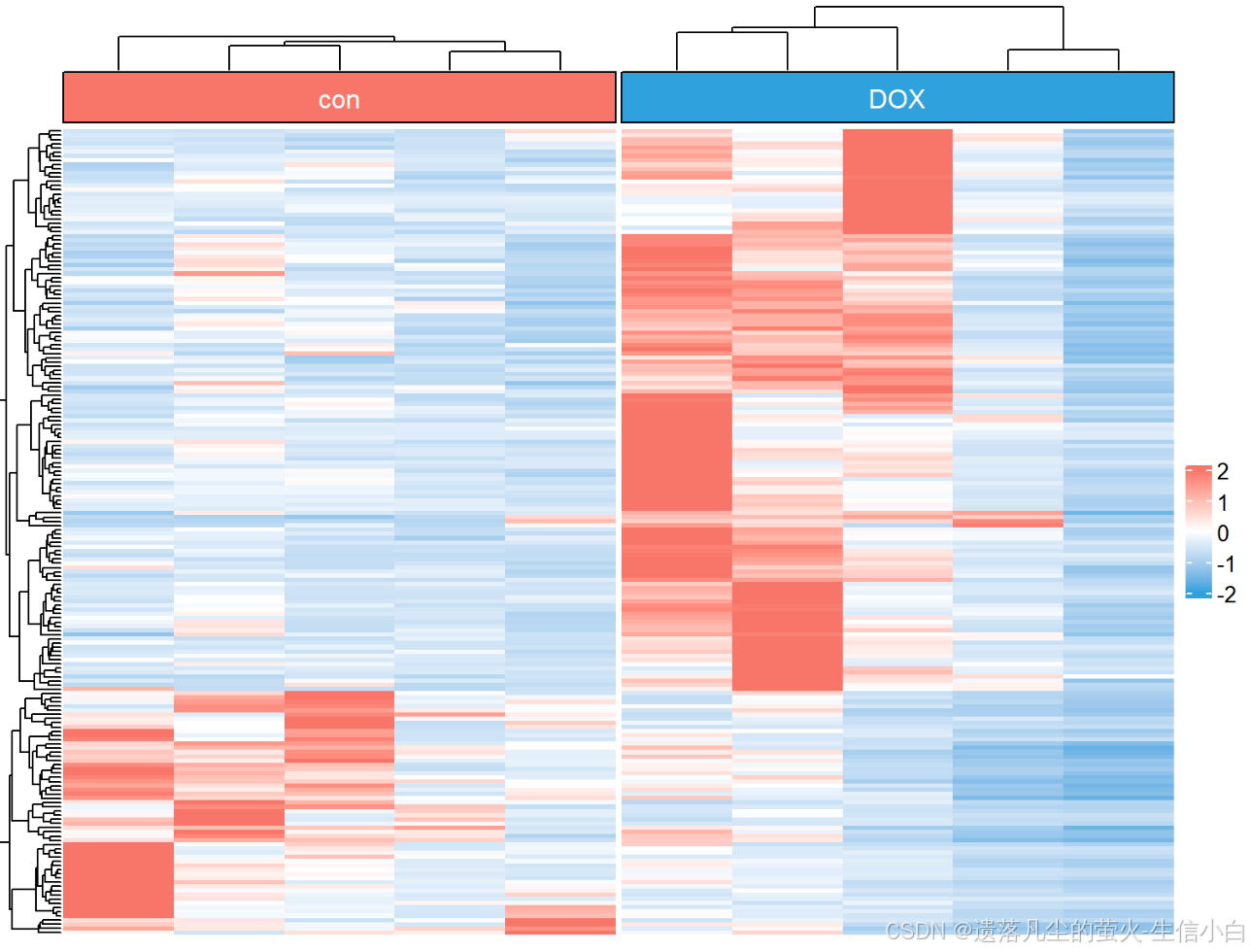
分组聚类的热图
library(ComplexHeatmap)
library(circlize)
col_fun = colorRamp2(c(-2, 0, 2), c("#2fa1dd", "white", "#f87669"))
top_annotation = HeatmapAnnotation(
cluster = anno_block(gp = gpar(fill = c("#f87669","#2fa1dd")),
labels = levels(Group),
labels_gp = gpar(col = "white", fontsize = 12)))
m = Heatmap(t(scale(t(exp[c(up,down),]))),name = " ",
col = col_fun,
top_annotation = top_annotation,
column_split = Group,
show_heatmap_legend = T,
border = F,
show_column_names = F,
show_row_names = F,
use_raster = F,
cluster_column_slices = F,
column_title = NULL)
m
示例三:此代码只适用于人的样本
打开Day11-5.20-case_2_GSE193861-geo_rnaseq_2
1.data
生信技能树
1.起个项目名字
TCGA的数据,统一叫TCGA-xxxx,非TCGA的数据随意起名,不要有特殊字符即可。
proj = "doxorubicin"
2.读取和整理数据
2.1 表达矩阵
library(tinyarray)
#> Warning: package 'tinyarray' was built under R version 4.3.3
get_count_txt("GSE193861")
dat = read.delim("GSE193861_raw_counts_GRCh38.p13_NCBI.tsv.gz",row.names = 1)
range(dat)
#> [1] 0 8150089
dat[1:4,1:4]
#> GSM5822748 GSM5822749 GSM5822750 GSM5822751
#> 100287102 3 2 1 2
#> 653635 53 39 15 45
#> 102466751 4 1 0 6
#> 107985730 0 0 0 1
# 转换为整数矩阵
exp = round(dat)
2.2 临床信息
library(tinyarray)
clinical = geo_download("GSE193861")$pd
#> Warning in geo_download("GSE193861"): exp is empty
3.表达矩阵行名ID转换
library(tinyarray)
exp = trans_entrezexp(exp)
#> Warning in bitr(rownames(entrezexp), fromType = "ENTREZID", toType = "SYMBOL",
#> : 4.17% of input gene IDs are fail to map...
exp[1:4,1:4]
#> GSM5822748 GSM5822749 GSM5822750 GSM5822751
#> DDX11L1 3 2 1 2
#> WASH7P 53 39 15 45
#> MIR6859-1 4 1 0 6
#> MIR1302-2HG 0 0 0 1
4.基因过滤
需要过滤一下那些在很多样本里表达量都为0或者表达量很低的基因。过滤标准不唯一。
过滤之前基因数量:
nrow(exp)
#> [1] 37734
常用过滤标准1:
仅去除在所有样本里表达量都为零的基因
exp1 = exp[rowSums(exp)>0,]
nrow(exp1)
#> [1] 30746
常用过滤标准2(推荐):
仅保留在一半以上样本里表达的基因
exp = exp[apply(exp, 1, function(x) sum(x > 0) > 0.5*ncol(exp)), ]
nrow(exp)
#> [1] 22415
5.分组信息获取
TCGA的数据,直接用make_tcga_group给样本分组(tumor和normal),其他地方的数据分组方式参考芯片数据pipeline/02_group_ids.R
p = identical(rownames(clinical),colnames(exp));p
#> [1] FALSE
if(!p) {
s = intersect(rownames(clinical),colnames(exp))
exp = exp[,s]
clinical = clinical[s,]
}
library(stringr)
#> Warning: package 'stringr' was built under R version 4.3.3
Group = str_split_i(clinical$title," ",6)
Group = factor(Group,levels = c("control","doxorubicin"))
table(Group)
#> Group
#> control doxorubicin
#> 5 4
6.保存数据
save(exp,Group,proj,clinical,file = paste0(proj,".Rdata"))
值得我奔走相告的神器:右键直接新建Rproj,挥泪告别setwd
规范统一格式的RNA-seq count及其标准化数据
基因长度并不是end-start