前言
在前面的章节中,我们介绍了如何使用 ggplot2 绘制热图
ggplot2 绘制热图的方式很多,如 geom_raster、geom_tile 等
但通常仅仅绘制热图是不够的,还需要对数据进行聚类,即绘制聚类热图。
例如,最常用的就是将差异基因的表达值绘制聚类热图,来查看基因在不同样本中的表达差异情况,或者比较不同聚类分组之间的差异。
绘制聚类热图的包有很多,我们主要介绍 pheatmap 和 ComplexHeatmap
简单示例
假设我们有如下数据
> df <- read.csv("~/Downloads/RPKM_DEG.csv", row.names = 1)
> df
control1 control2 control3 treat1 treat2 treat3
Gene1 178.413 180.616 111.951 44.264 44.251 35.842
Gene2 1790.491 33.799 3076.195 533.618 527.694 493.286
Gene3 55.313 72.512 42.625 26.291 31.324 26.485
Gene4 138.170 10.234 168.461 1014.917 968.487 1013.409
Gene5 400.783 417.081 422.252 16.958 21.532 18.350
Gene6 16.466 10.856 17.788 117.155 113.056 117.383
Gene7 282.103 484.187 192.498 73.769 68.207 72.967
...
要绘制简单的热图,可以使用内置的 heatmap 函数
heatmap(as.matrix(df))
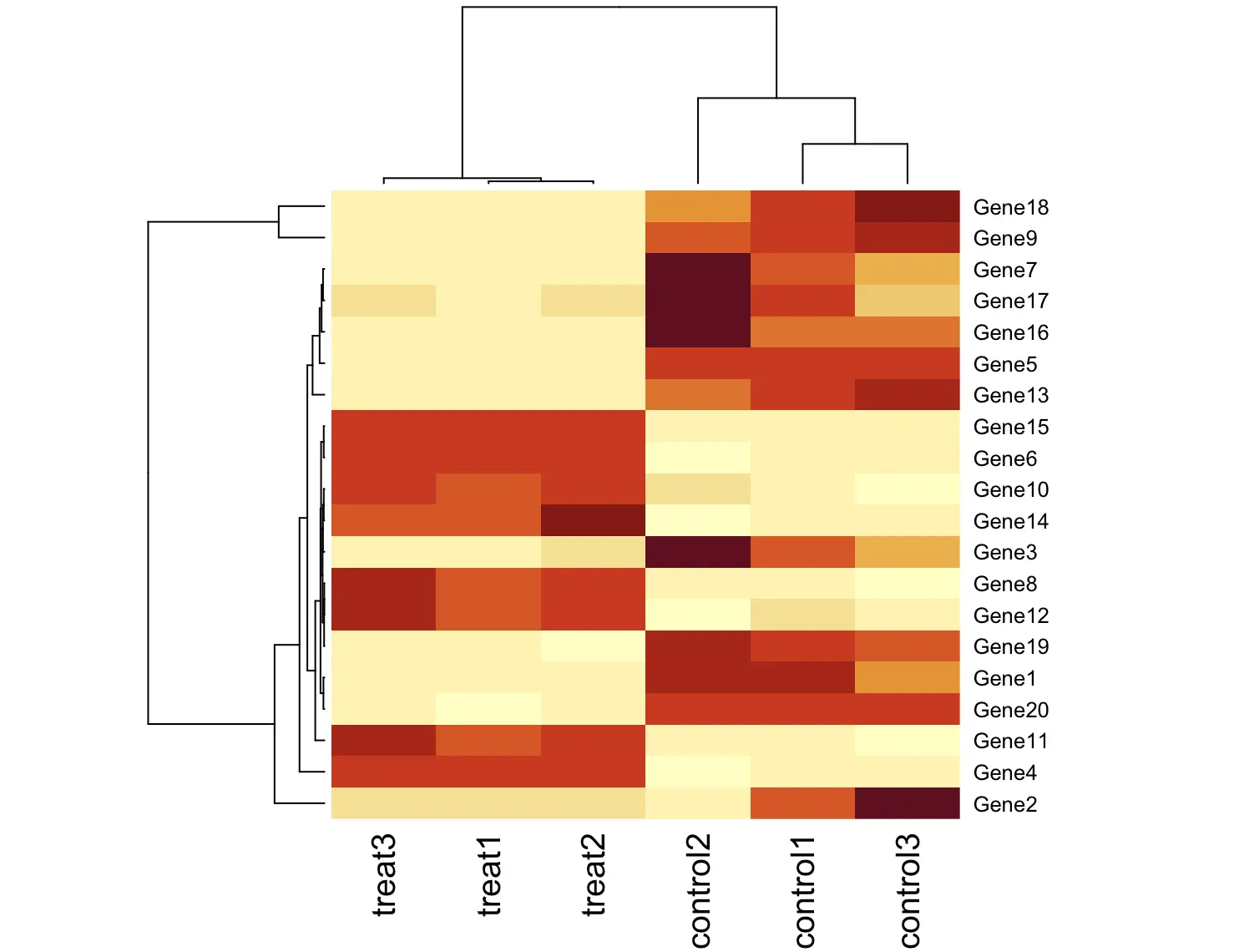
更改颜色,并为列添加列样本的分类颜色条
heatmap(as.matrix(df), col = rainbow(10)[3:9], ColSideColors = rep(c("pink", "purple"), each = 3))
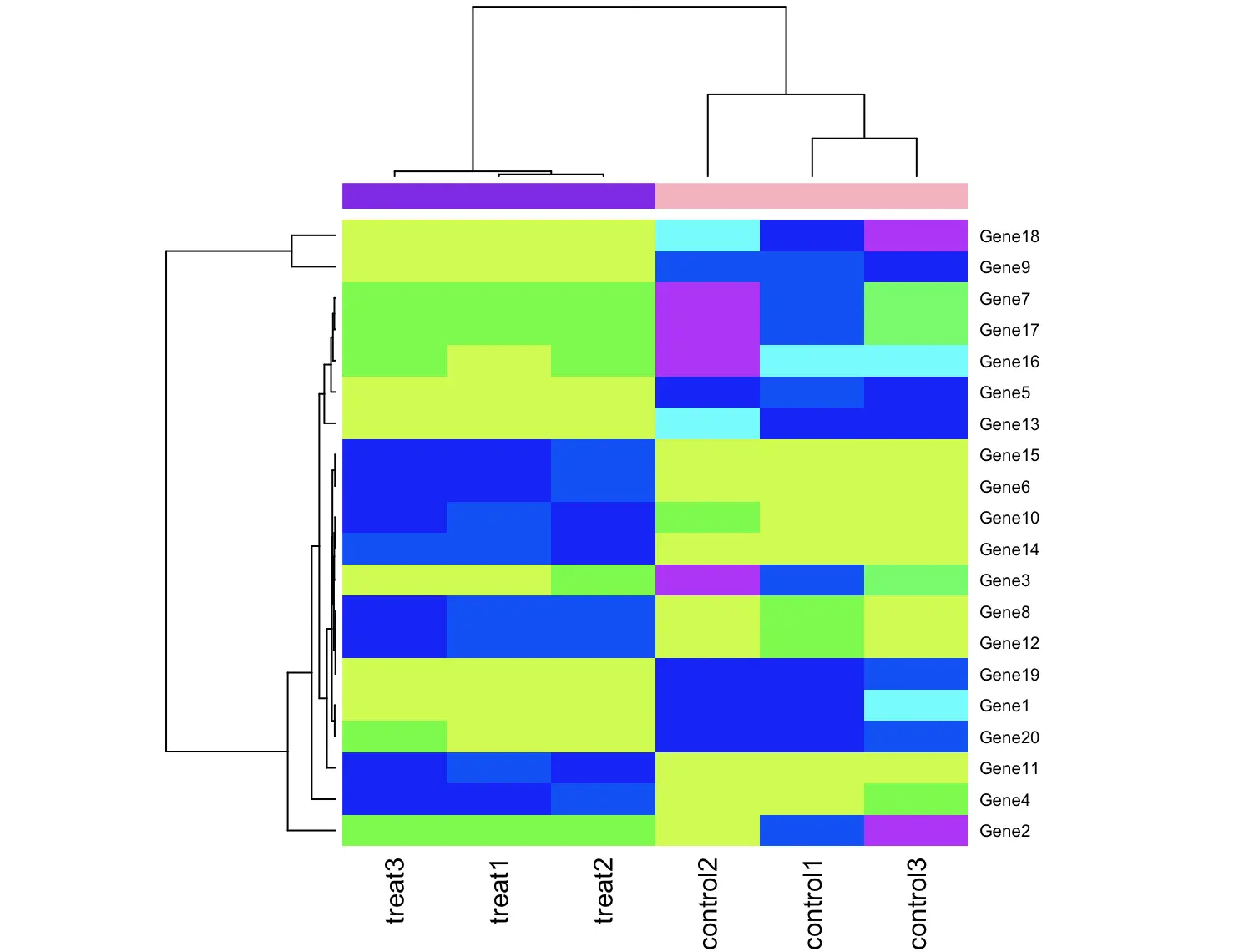
内置函数提供的样式较少,无法对某些图形属性进行设置。
所以下面我们使用 pheatmap 包来绘制热图
pheatmap
pheatmap 对图形属性提供了更精细的控制
1. 安装导入
install.packages("pheatmap")
library(pheatmap)
2. 简单示例
pheatmap(df)
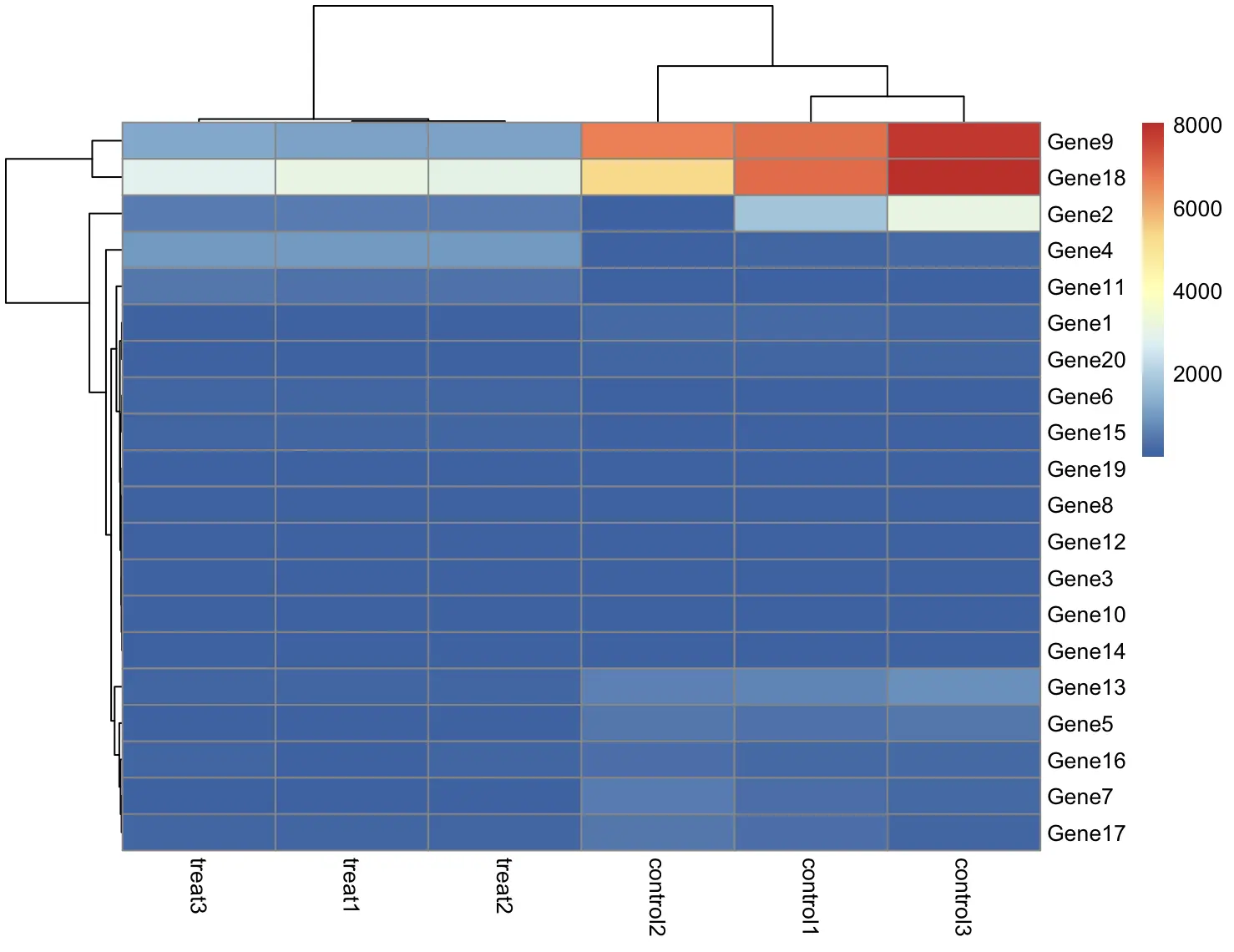
这样看起来怪怪的,应该是基因的表达量差异,所以对行进行标准化
pheatmap(df, scale = "row")
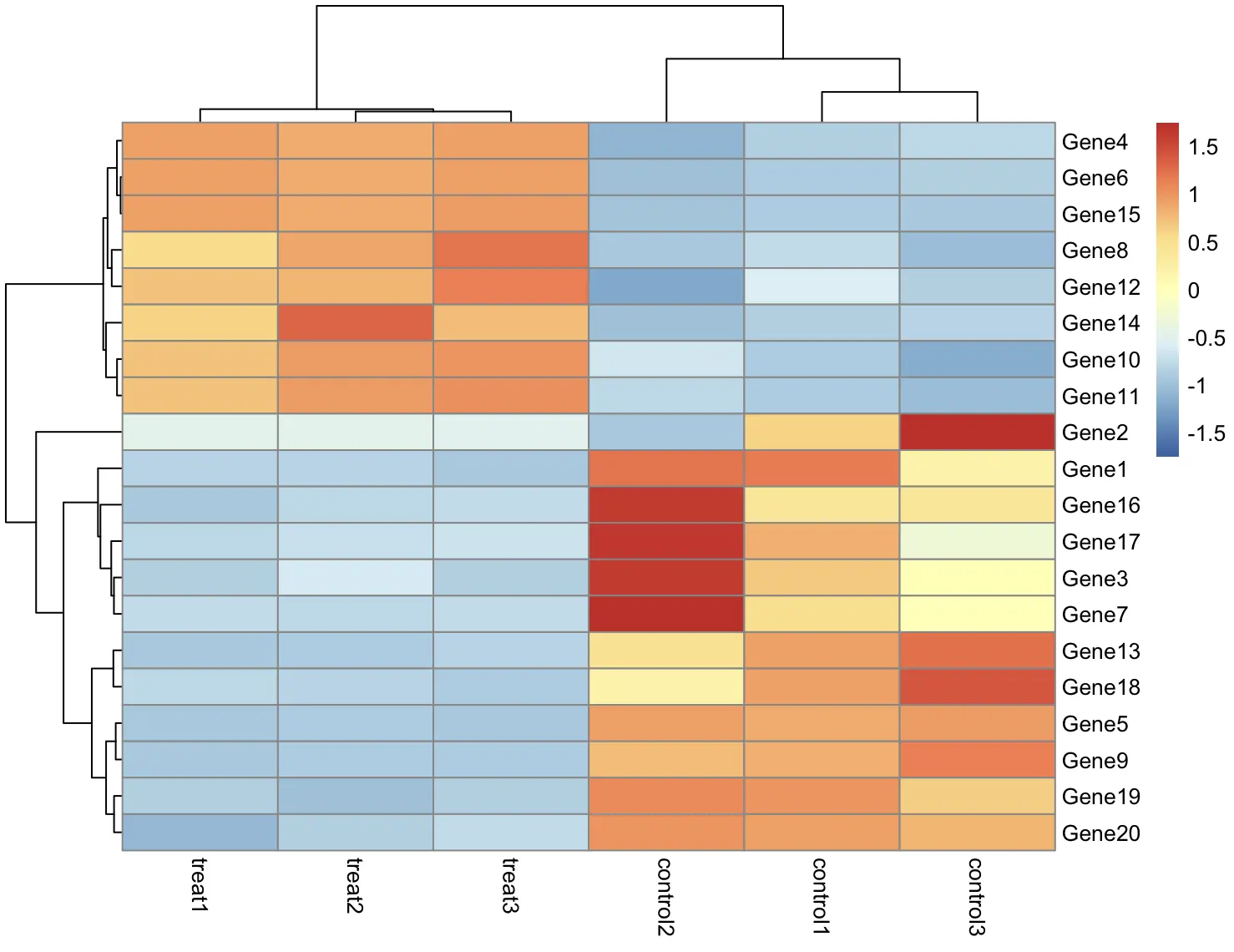
嗯,一下子就顺眼多了,实验组和对照组的基因表达量差别明显
3. 聚类
默认情况下,会对数据的行列分别进行层次聚类,如果我们想在进行层次聚类之前,先对行特征,也就是基因进行 k-means 聚类,我们可以
pheatmap(df, scale = "row", kmeans_k = 3)
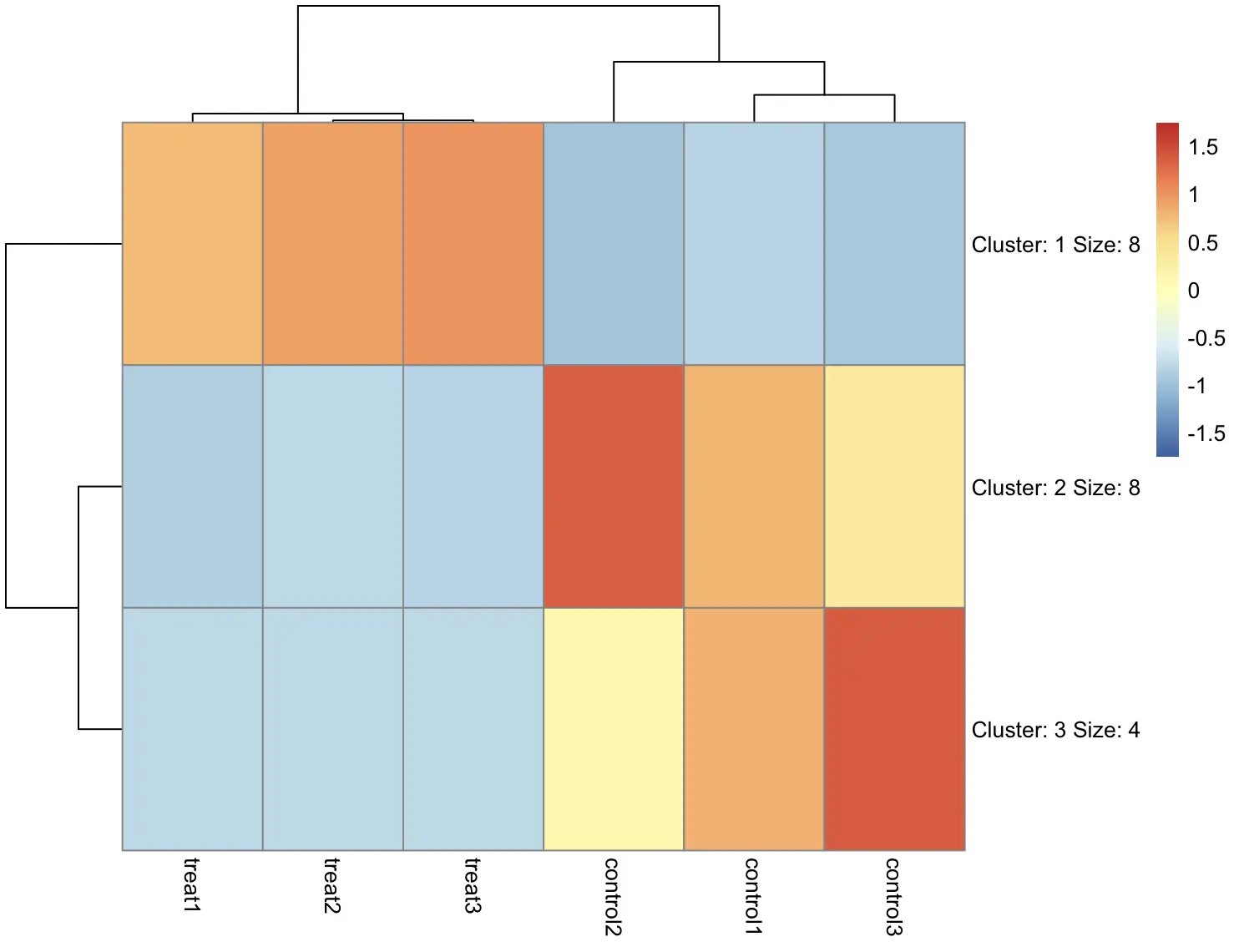
先将基因聚为 3 类,再进行层次聚类
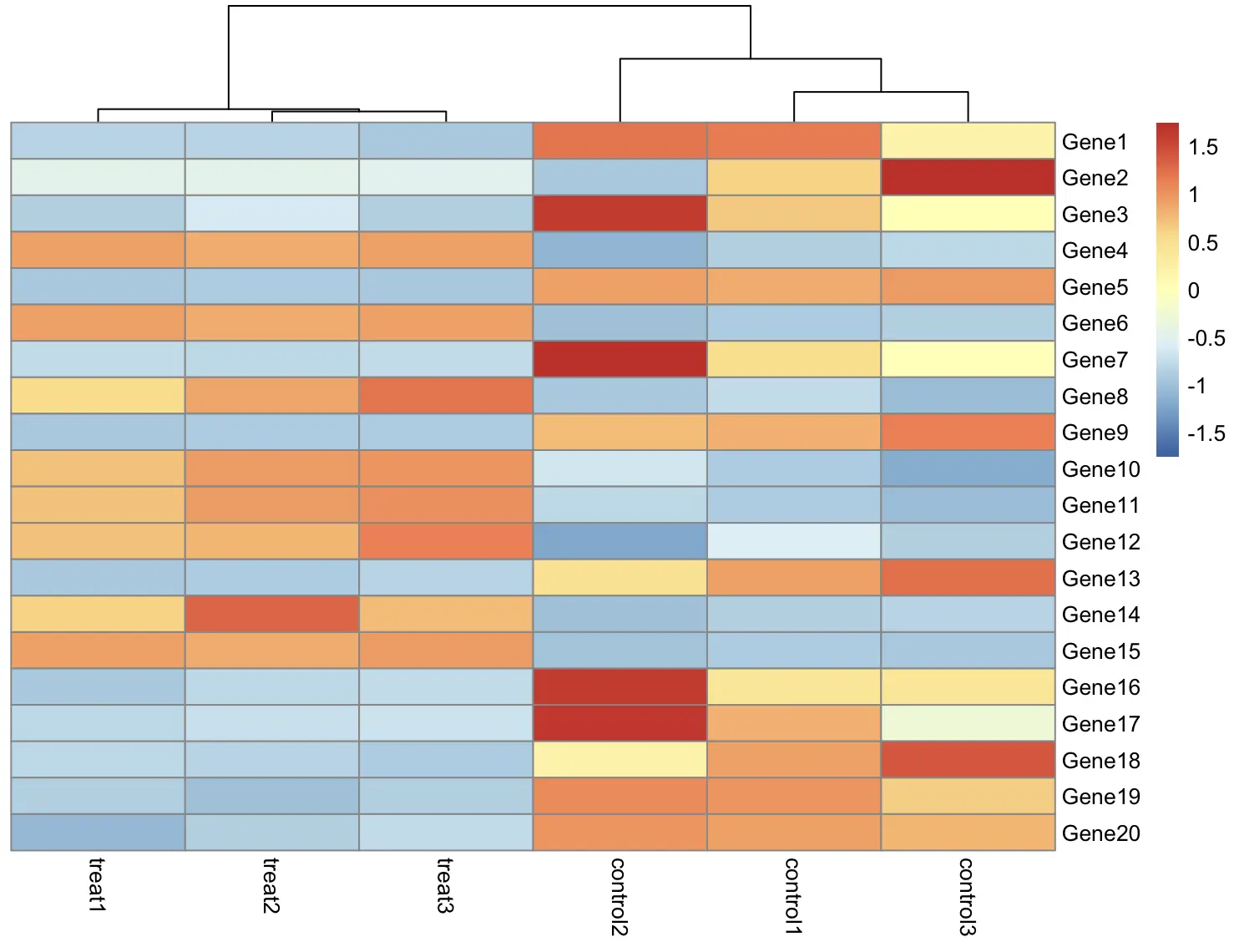
如果只想对其中行列中的一个进行聚类,可以使用 cluster_rows 和 cluster_cols 参数,取消对行或列的距离
pheatmap(df, scale = "row", cluster_rows = FALSE)
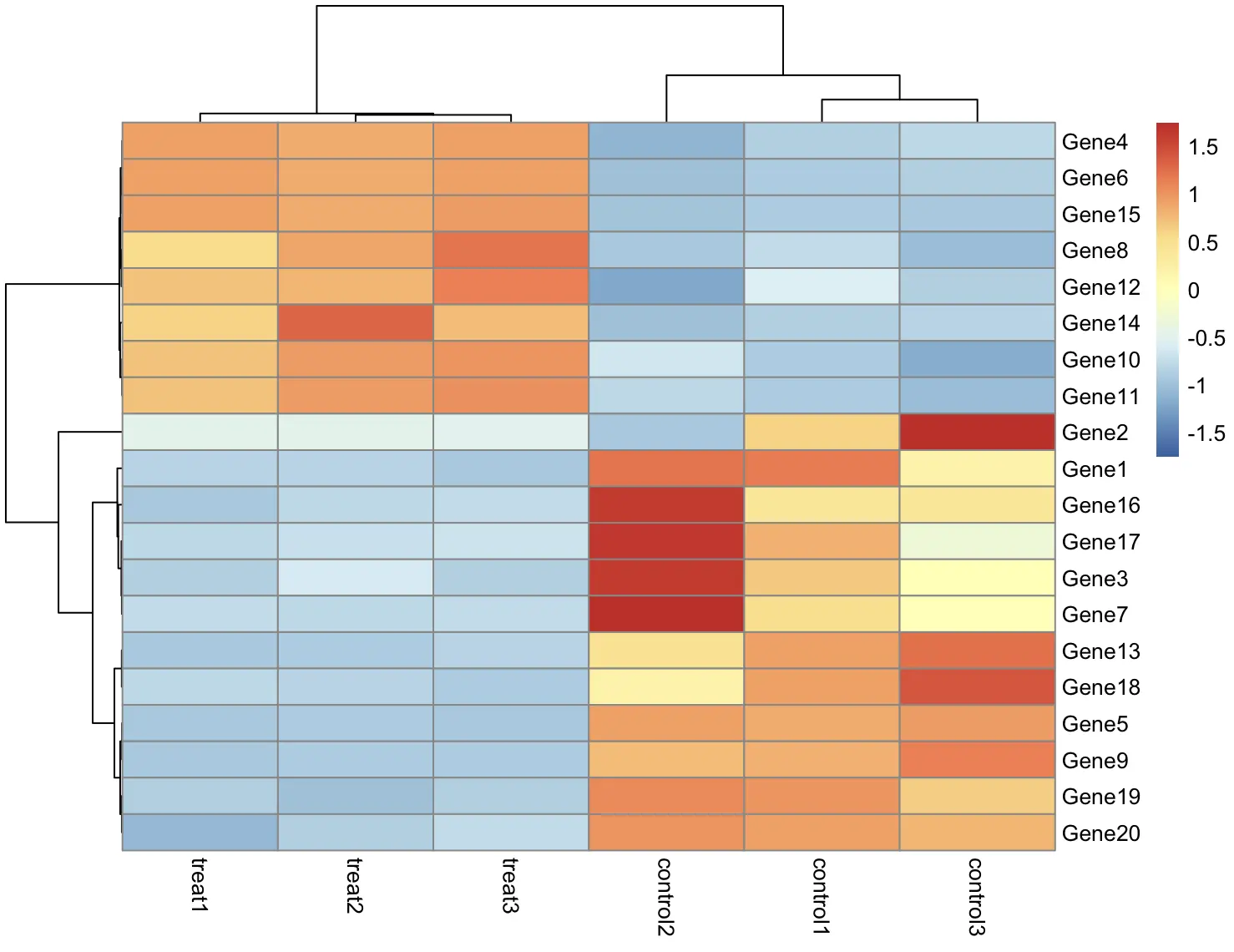
默认的距离度量为欧氏距离,也可以分别为行列指定不同的距离度量,例如
pheatmap(df, scale = "row", clustering_distance_rows = "correlation", clustering_distance_cols = "manhattan")
也可以使用 clustering_method 参数来指定不同的聚类方法,支持以下几种方法:
'ward', 'ward.D', 'ward.D2',
'single', 'complete', 'average',
'mcquitty', 'median', 'centroid'
pheatmap(df, scale = "row", clustering_method = "ward.D")
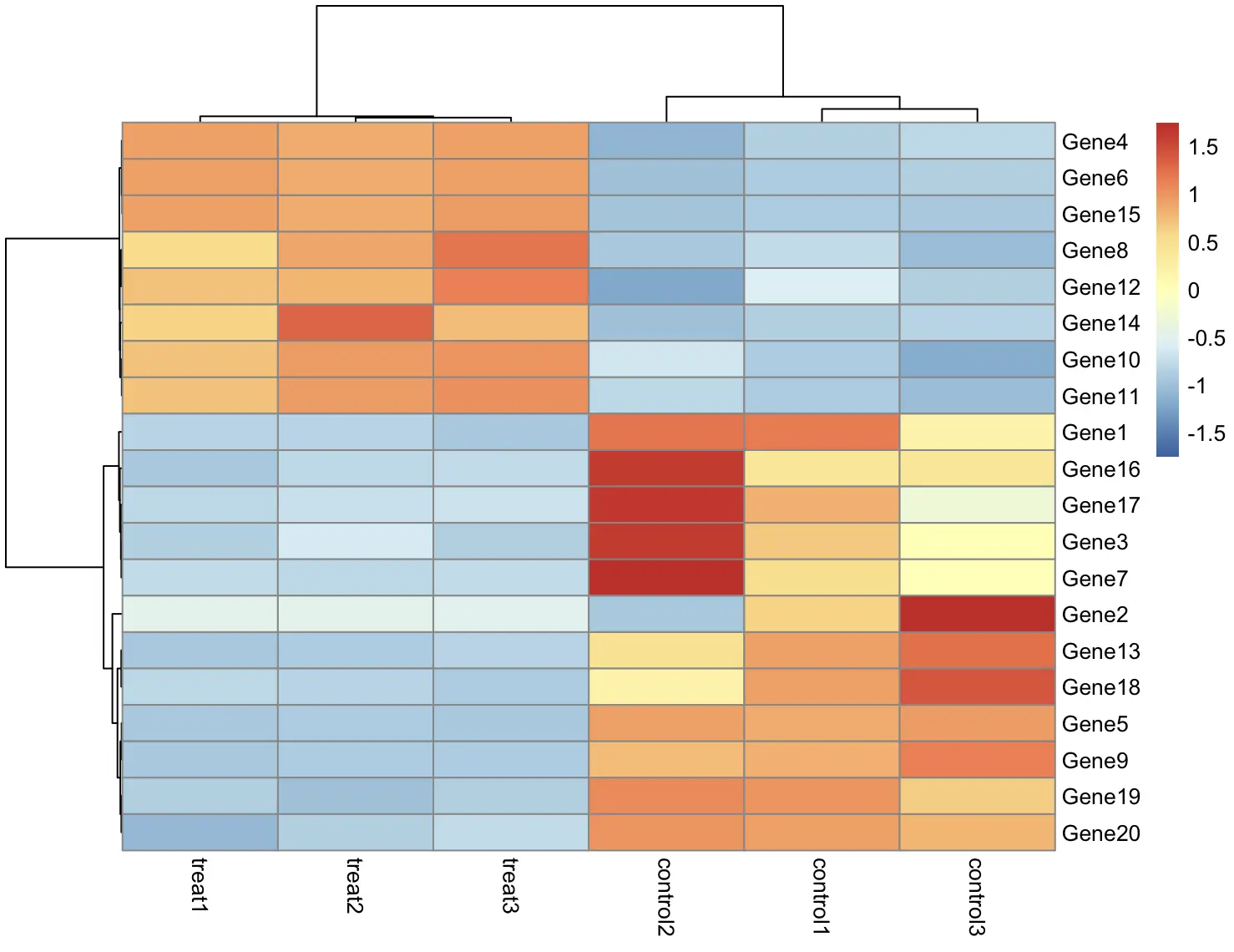
4. 图例
图例的设置很简单,即通过 legend_breaks 参数设置断点,legend_labels 参数设置断点处的标签
pheatmap(df, scale = "row", legend_breaks = c(-1, 0, 1), legend_labels = c("low", "median", "high"))
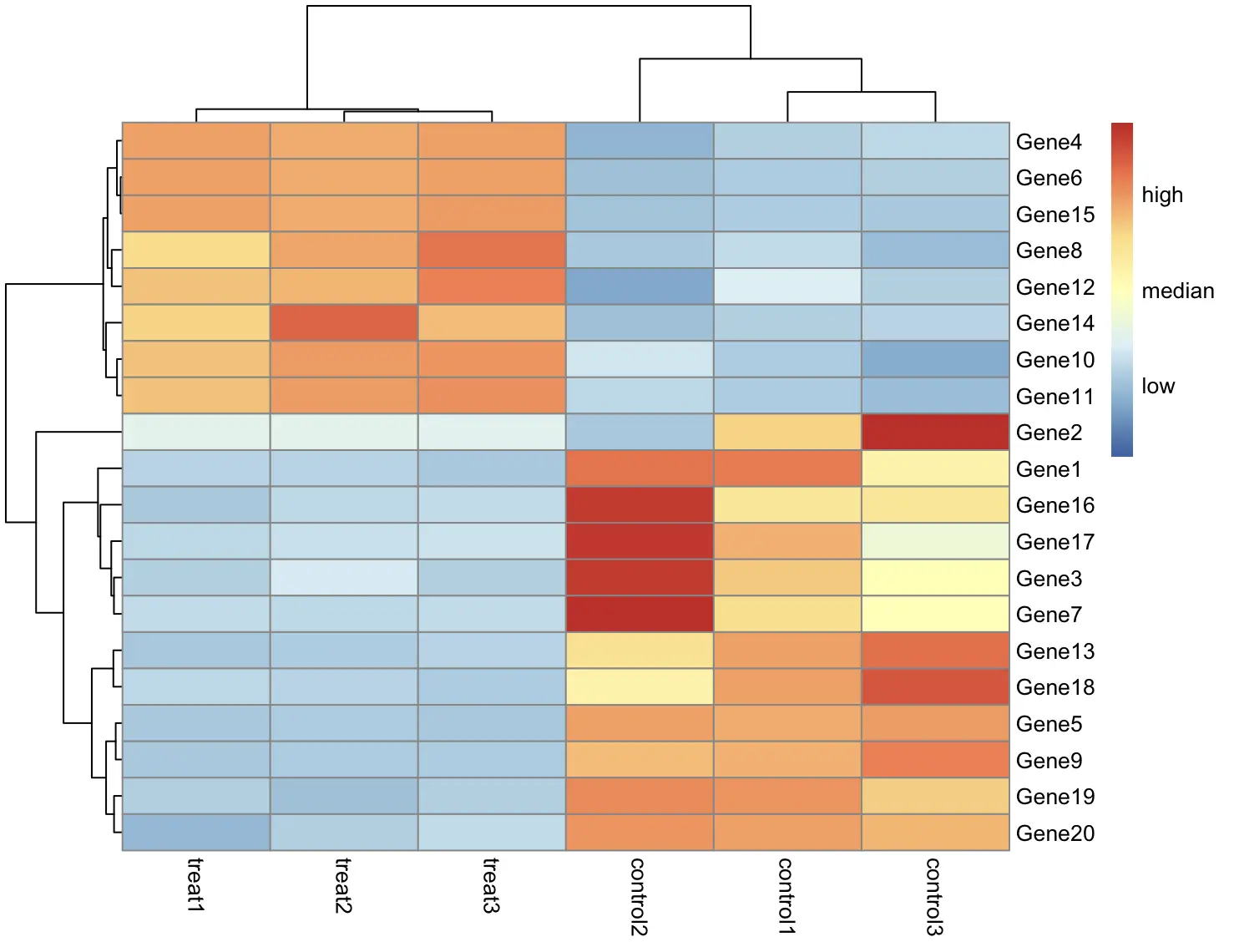
如果不想显示图例,直接设置 legend = F 就行
5. 边框
设置边框颜色
pheatmap(df, scale = "row", border_color = "white")
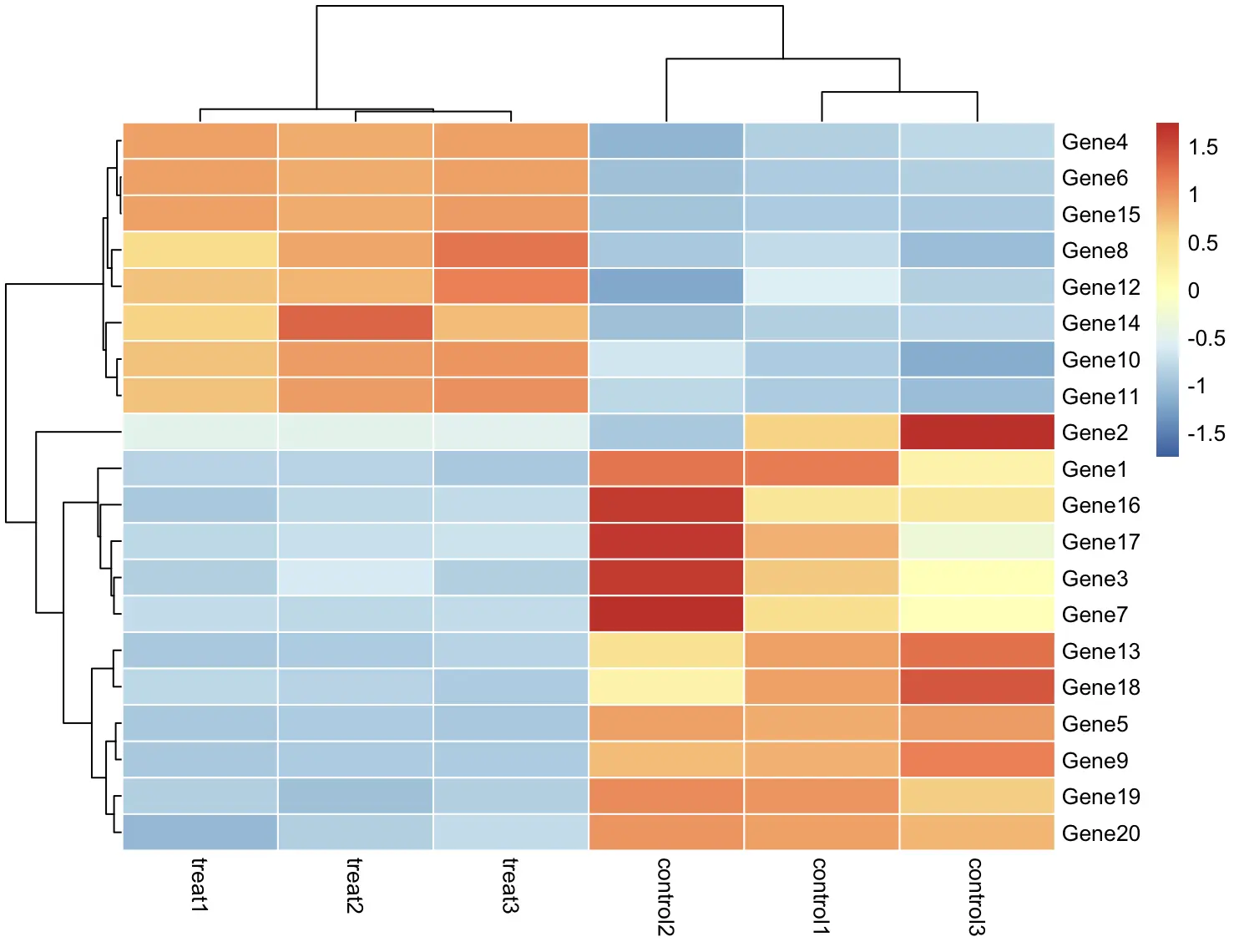
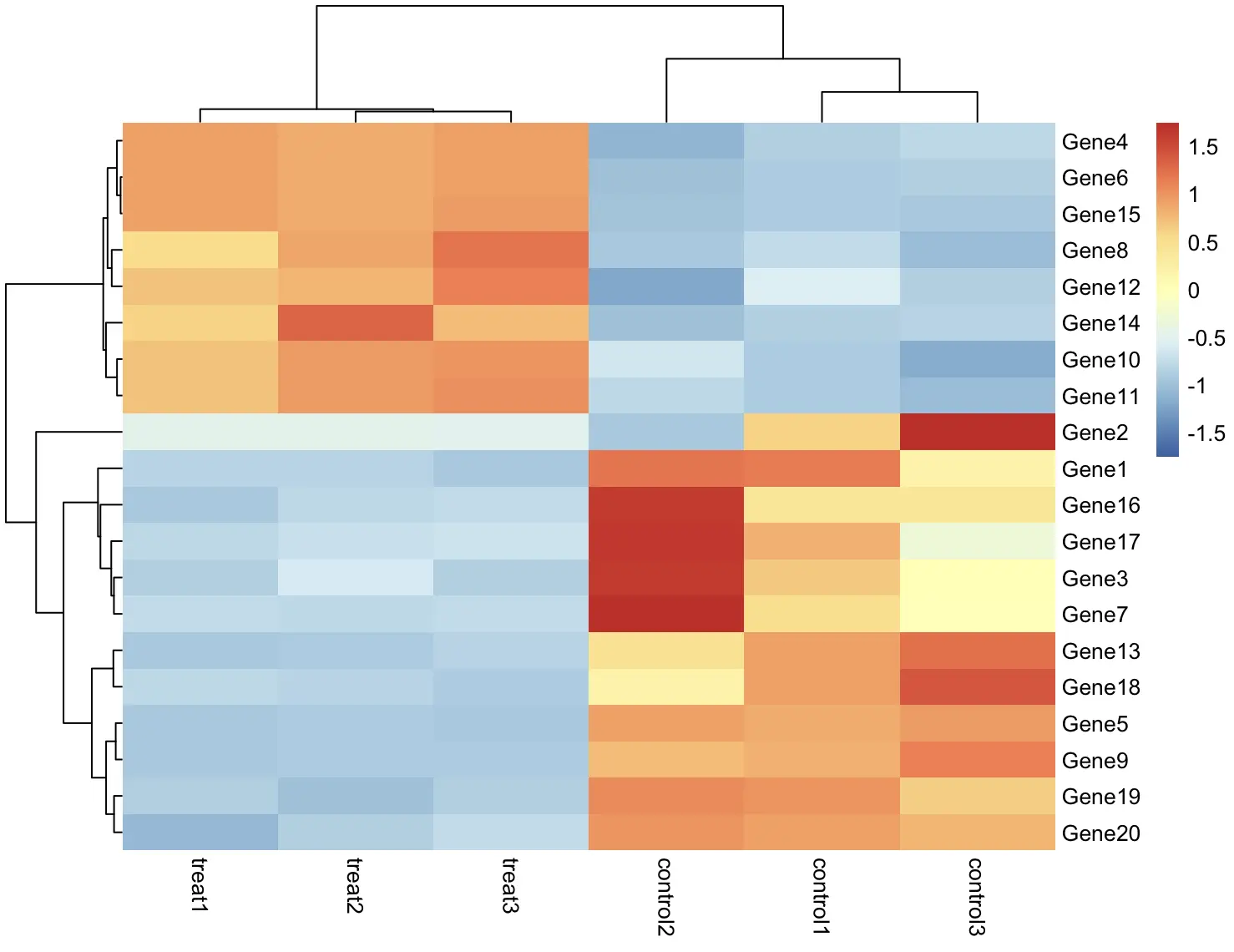
删除边框
pheatmap(df, scale = "row", border = F)
6. 单元格
默认情况下,单元格的长度和宽度会根据图片的大小自动调整,如果想固定单元格的大小,可以使用 cellwidth 和 cellheight 两个参数
pheatmap(df, scale = "row", cellwidth = 15, cellheight = 15)
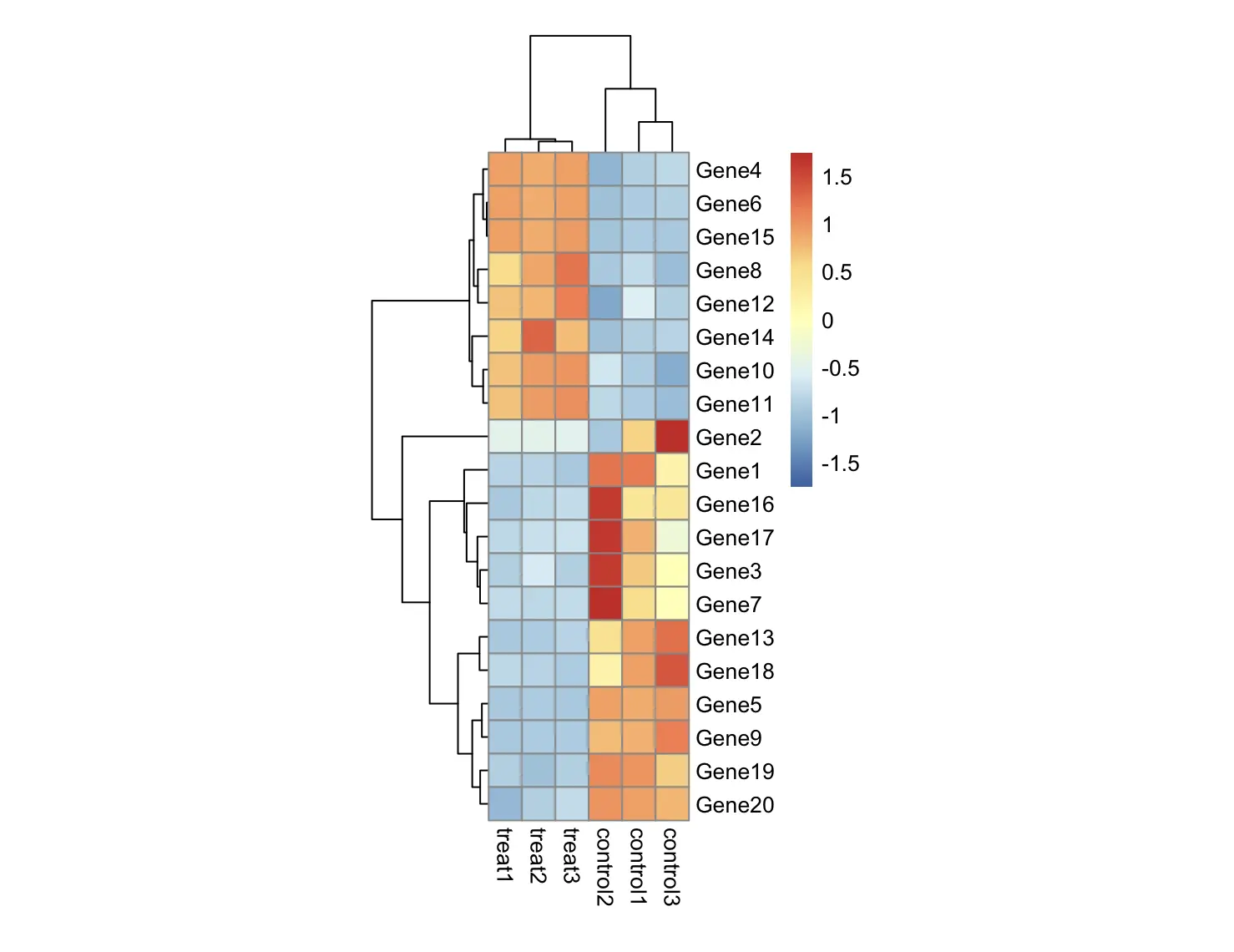
如果我们想在单元格中显示对于的数值,可以设置 display_numbers = TRUE
pheatmap(df, scale = "row", display_numbers = TRUE)
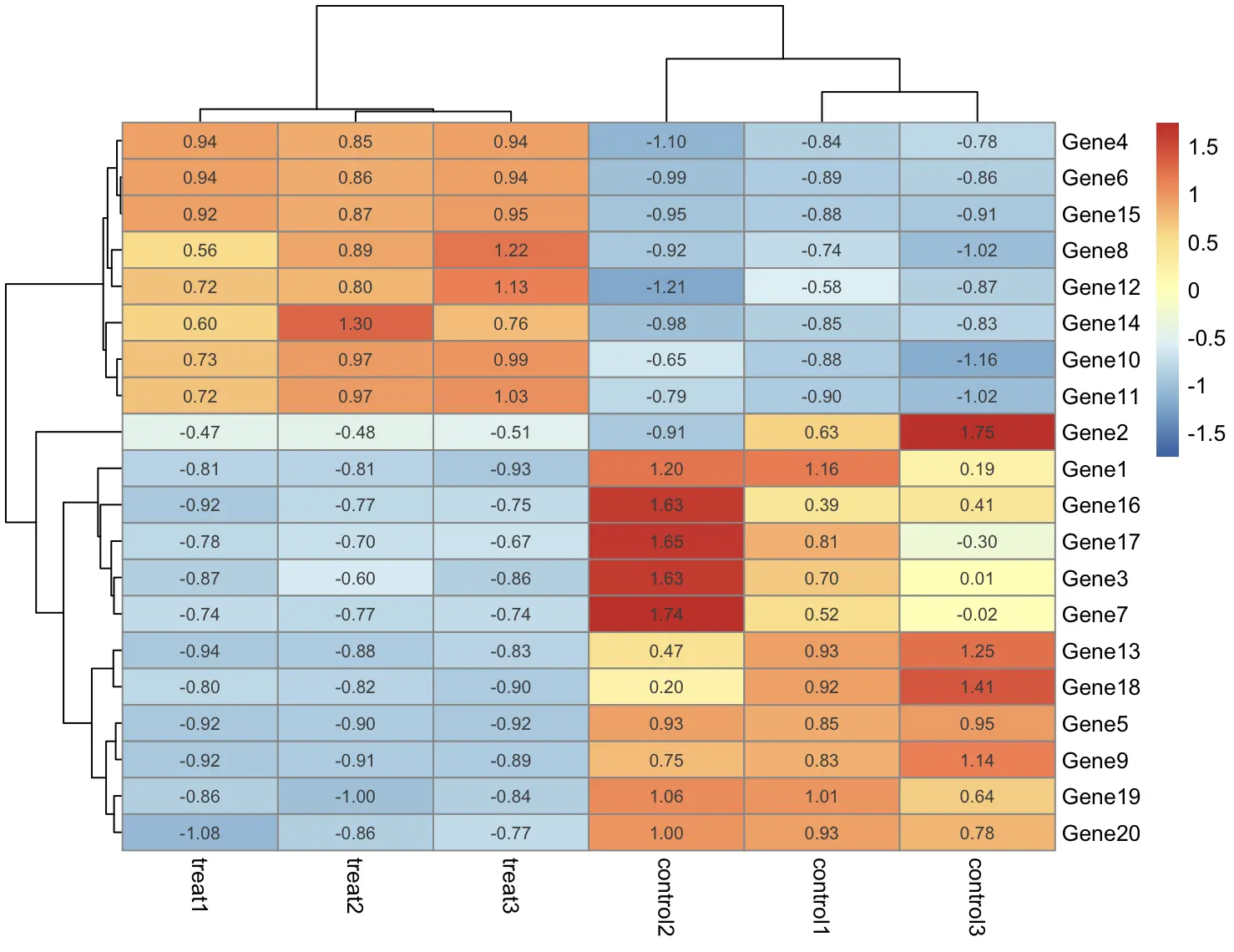
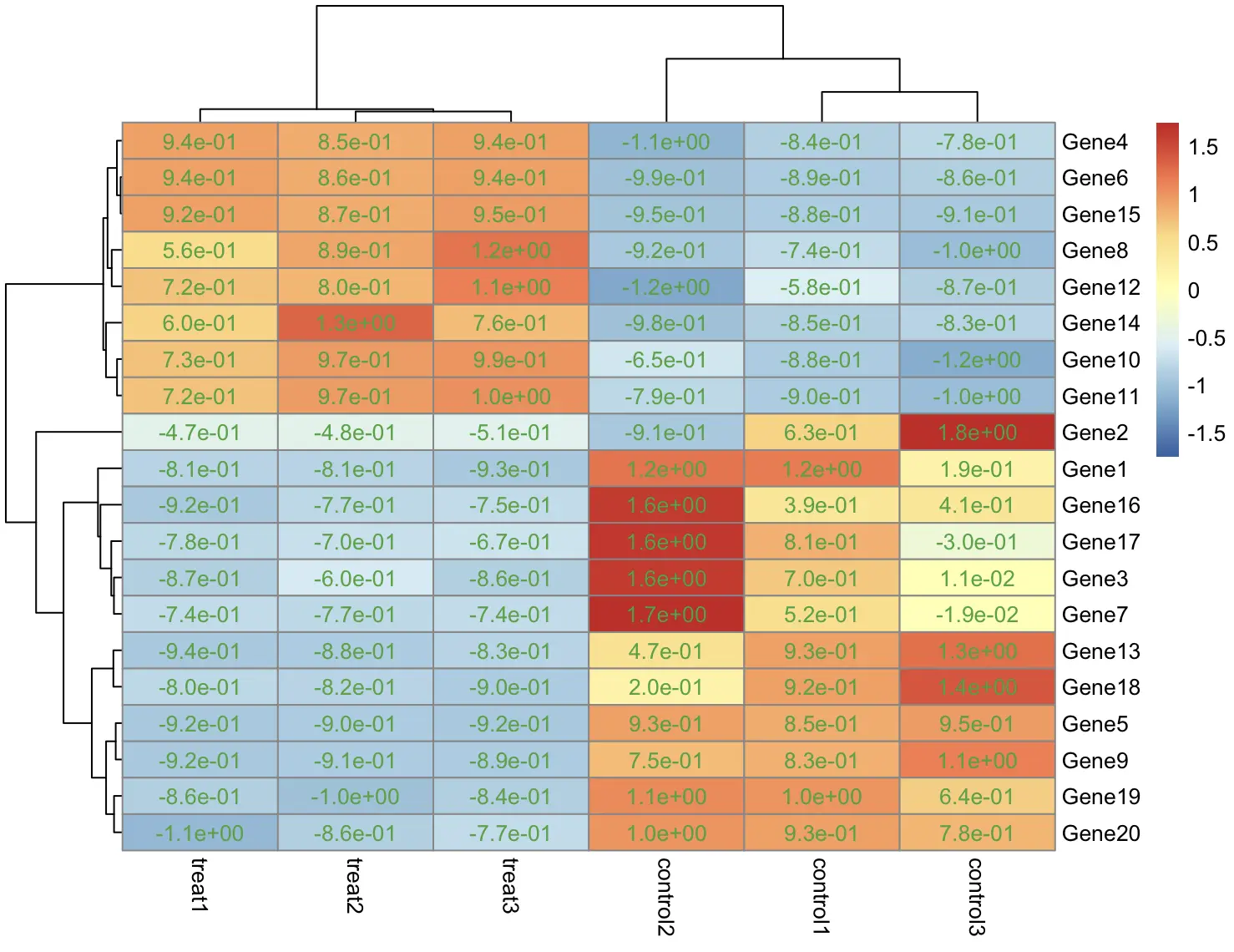
对显示的数值进行格式化
pheatmap(
df,
scale = "row",
display_numbers = TRUE,
# 显示为科学计数法
number_format = "%.1e",
# 设置颜色
number_color = "#4daf4a",
# 设置数值字体大小
fontsize_number = 10
)
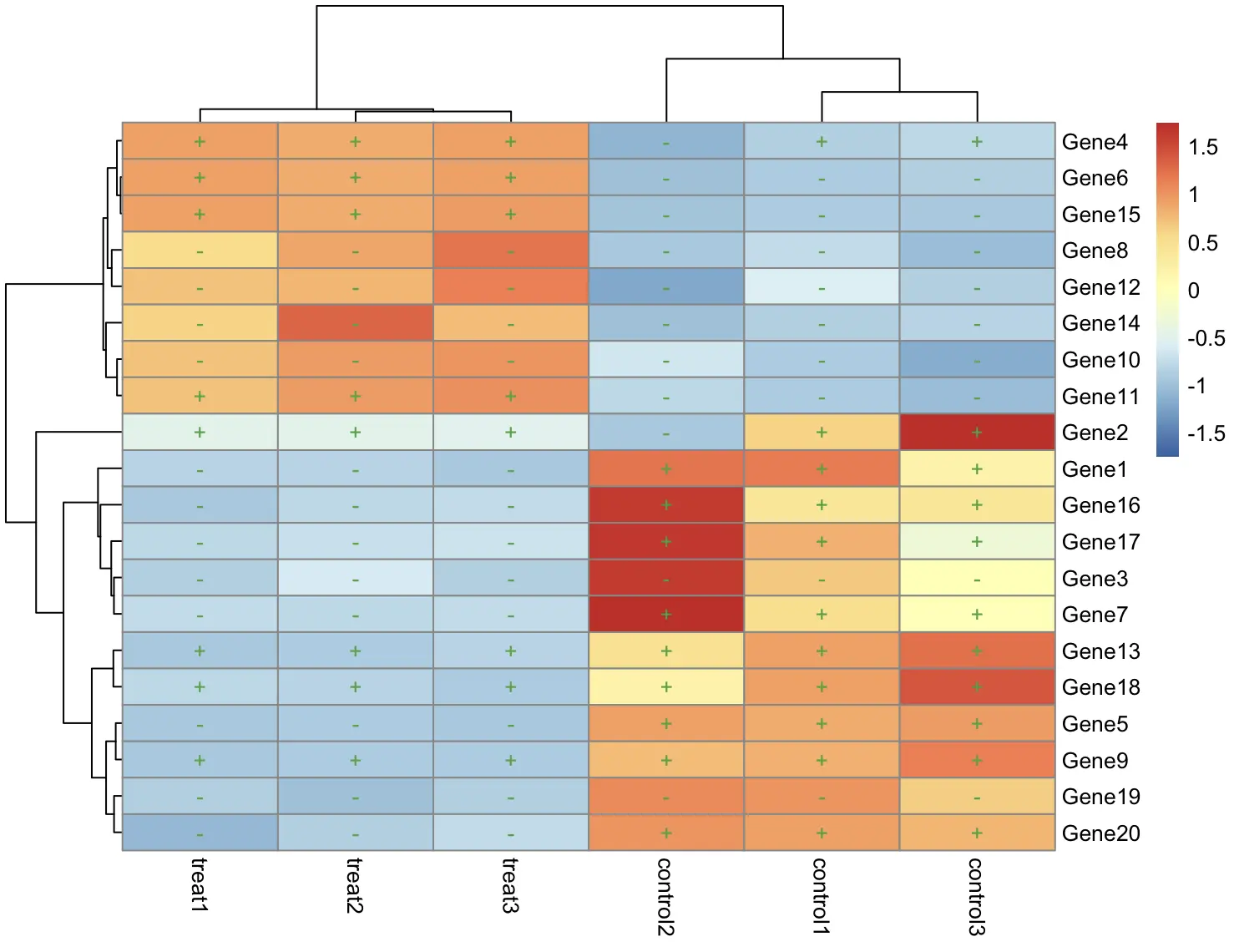
或者,为 display_numbers 参数传递一个矩阵
例如,根据表达值是否大于 100 来显示不同的标记
pheatmap(
df, scale = "row",
display_numbers = matrix(ifelse(df > 100, "+", "-"), nrow = nrow(df)),
number_color = "#4daf4a",
fontsize_number = 10
)
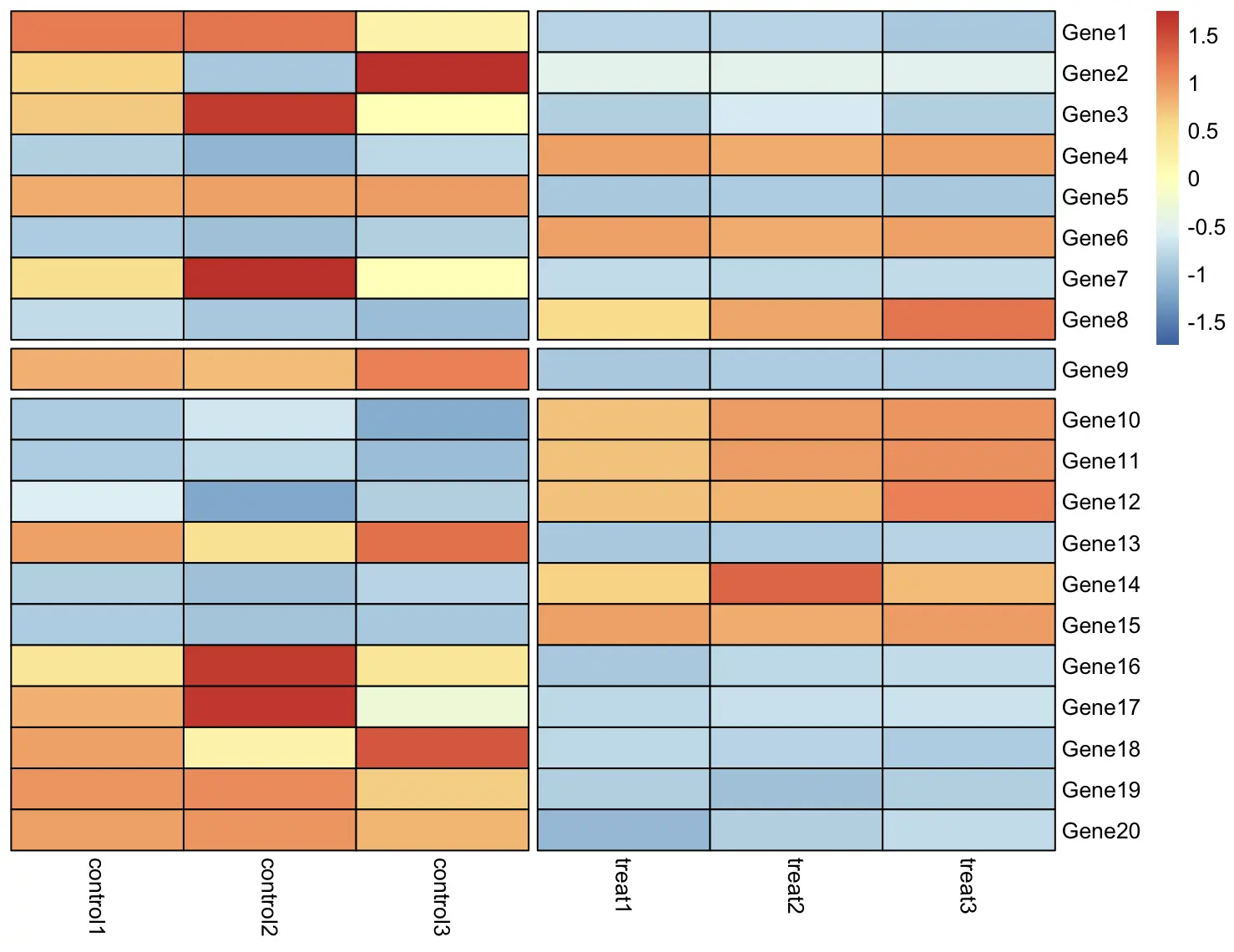
7. 分块
在不对数据进行聚类的情况下,可以对行列进行自定义划分为不同的块
pheatmap(
df,
scale = "row",
cluster_rows = FALSE,
cluster_cols = FALSE,
gaps_col = 3,
gaps_row = c(8, 9),
border_color = "black"
)
或者只对行或列进行分块
pheatmap(
df,
scale = "row",
cluster_rows = FALSE,
gaps_row = c(8, 12, 16),
border_color = "black"
)
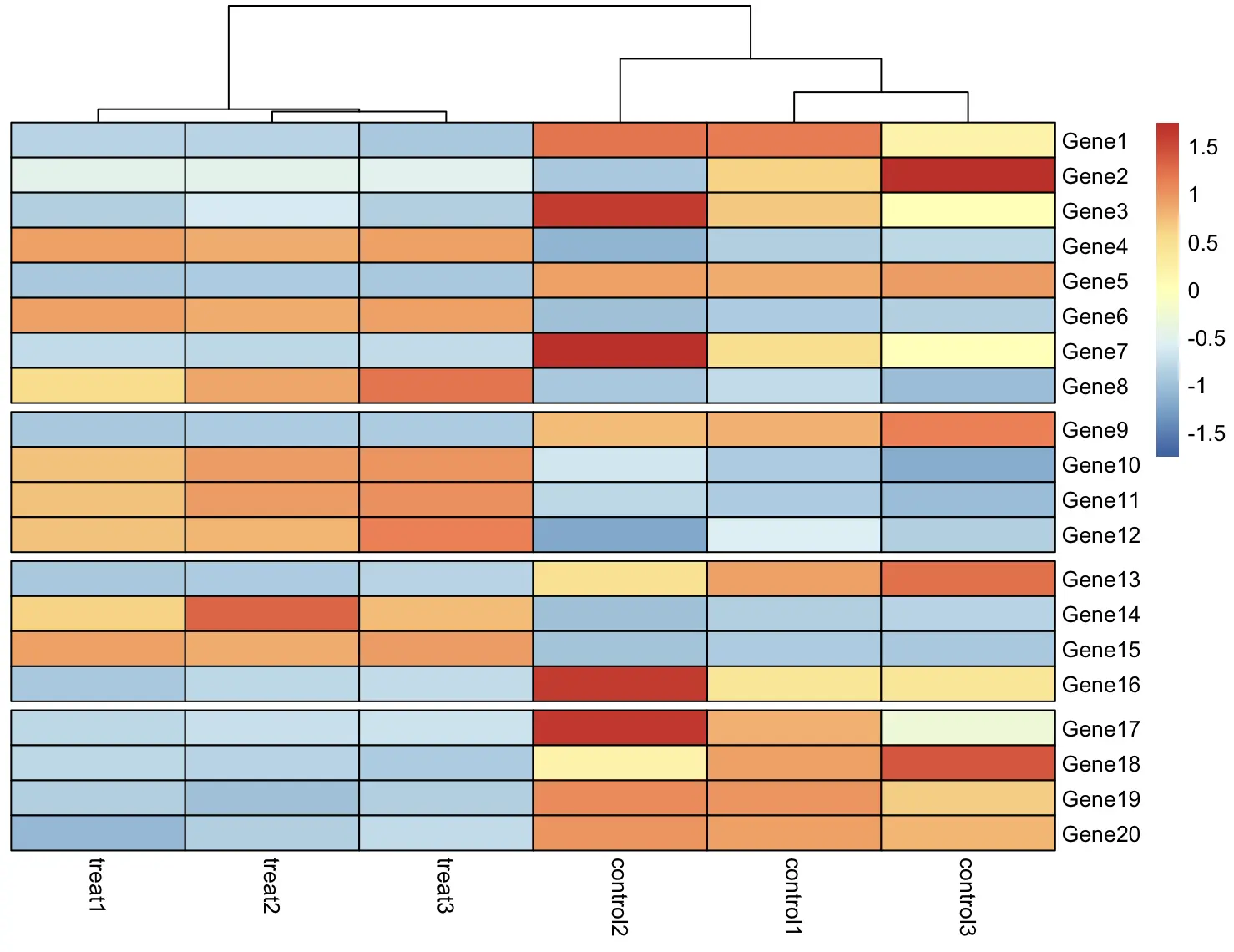
总之,只能对未聚类的行或列进行分块
或者,根据层次聚类的结果,对数据进行分块
pheatmap(
df,
scale = "row",
cutree_rows = 3,
cutree_cols = 2
)
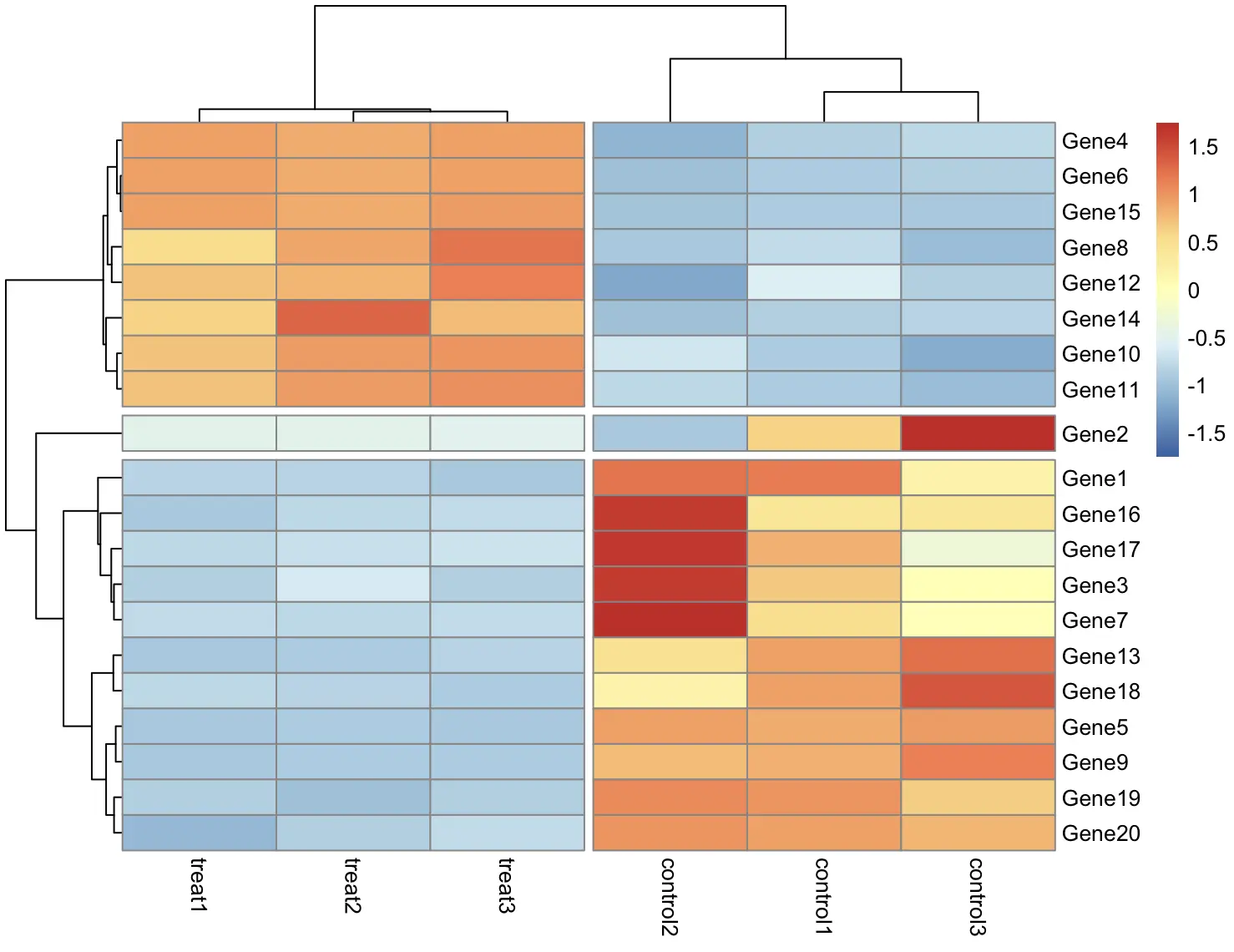
8. 标签
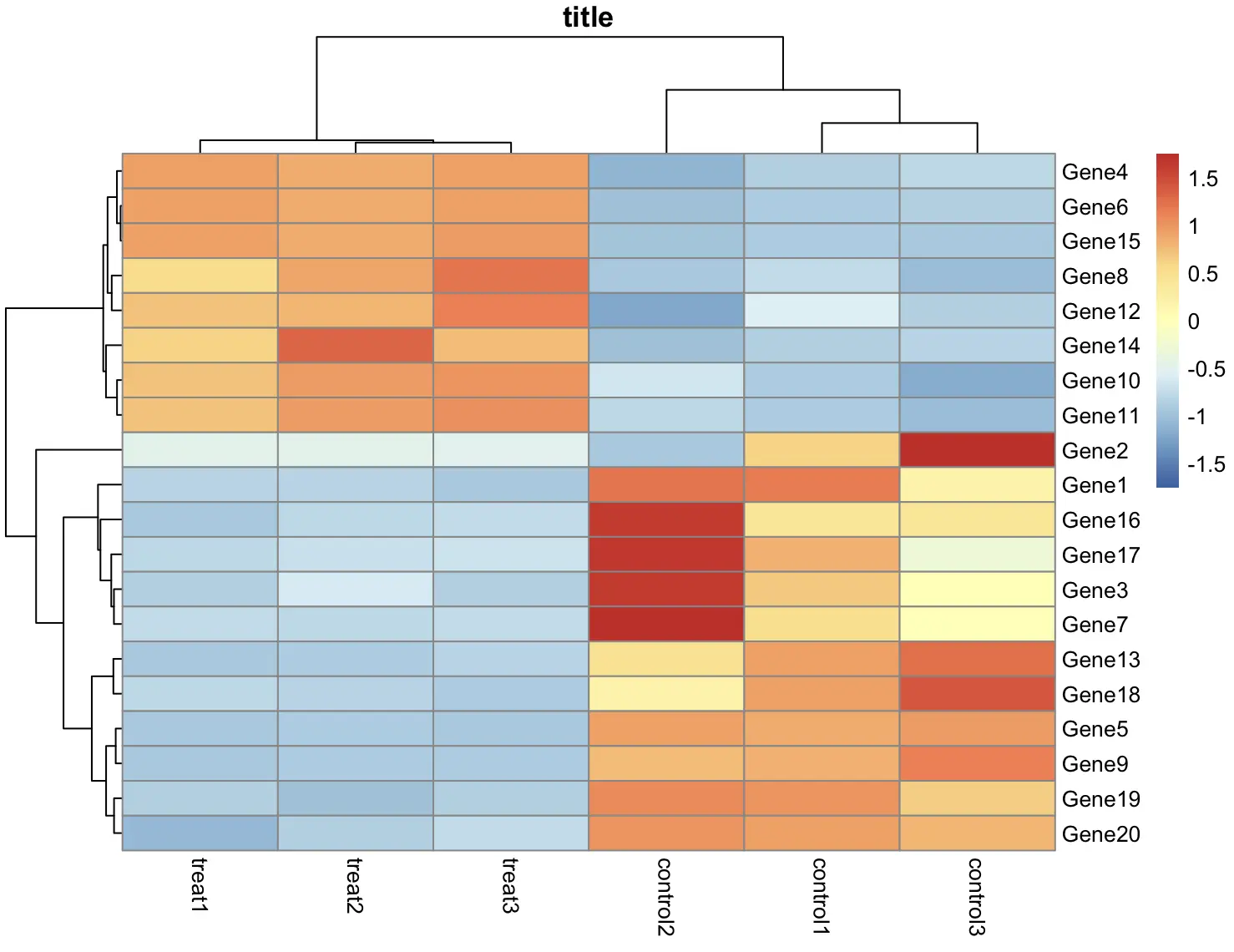
使用 main 参数来设置图像的标题
pheatmap(
df, scale = "row",
main = "title"
)
可以使用 show_colnames 和 show_rownames 不显示标签
pheatmap(
df, scale = "row",
show_colnames = FALSE,
show_rownames = FALSE
)
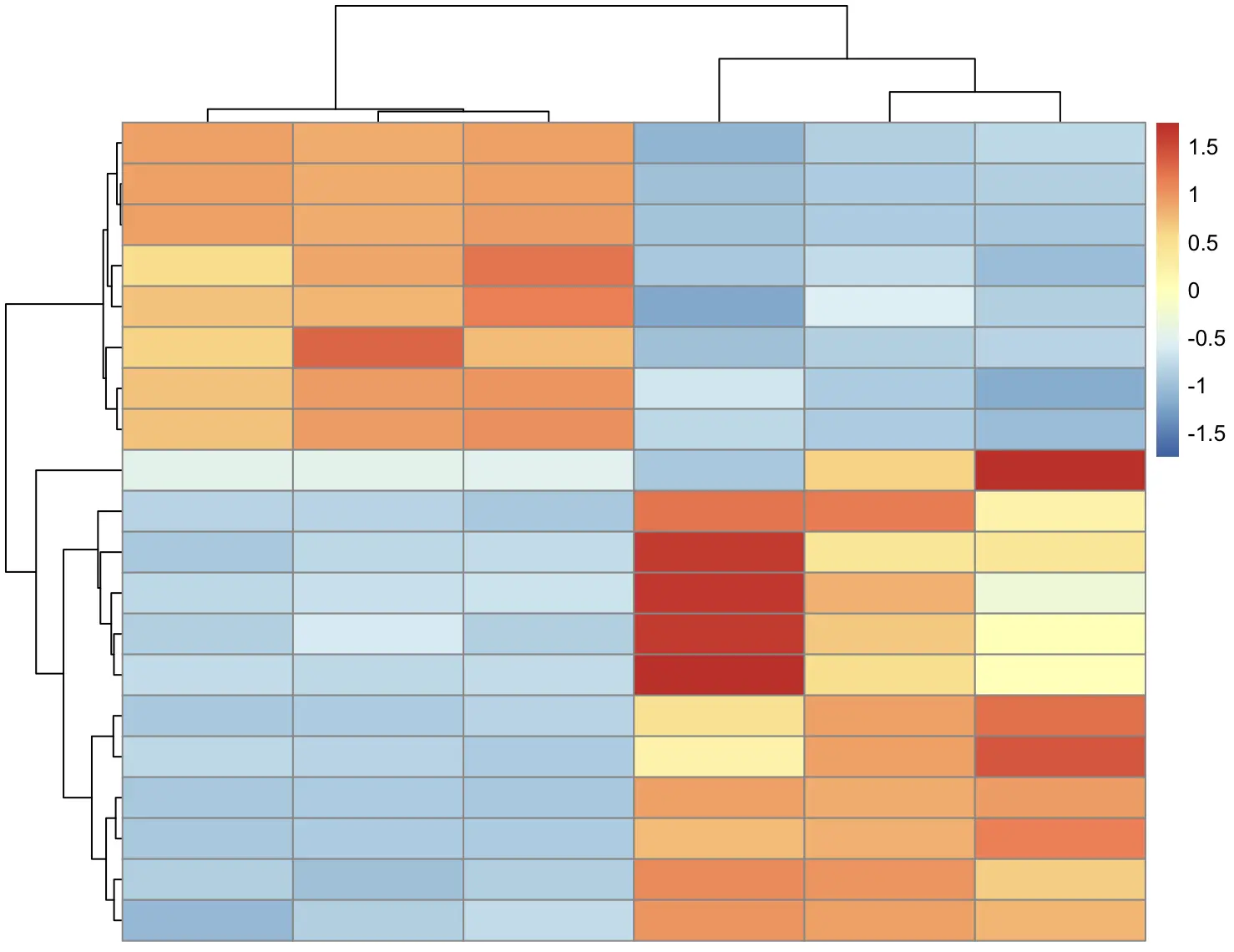
分别设置标签的大小,同时设置列标签的倾斜角度,可选的角度有 270、0、45、90、315
pheatmap(
df, scale = "row",
fontsize_row = 8,
fontsize_col = 12,
angle_col = 45
)
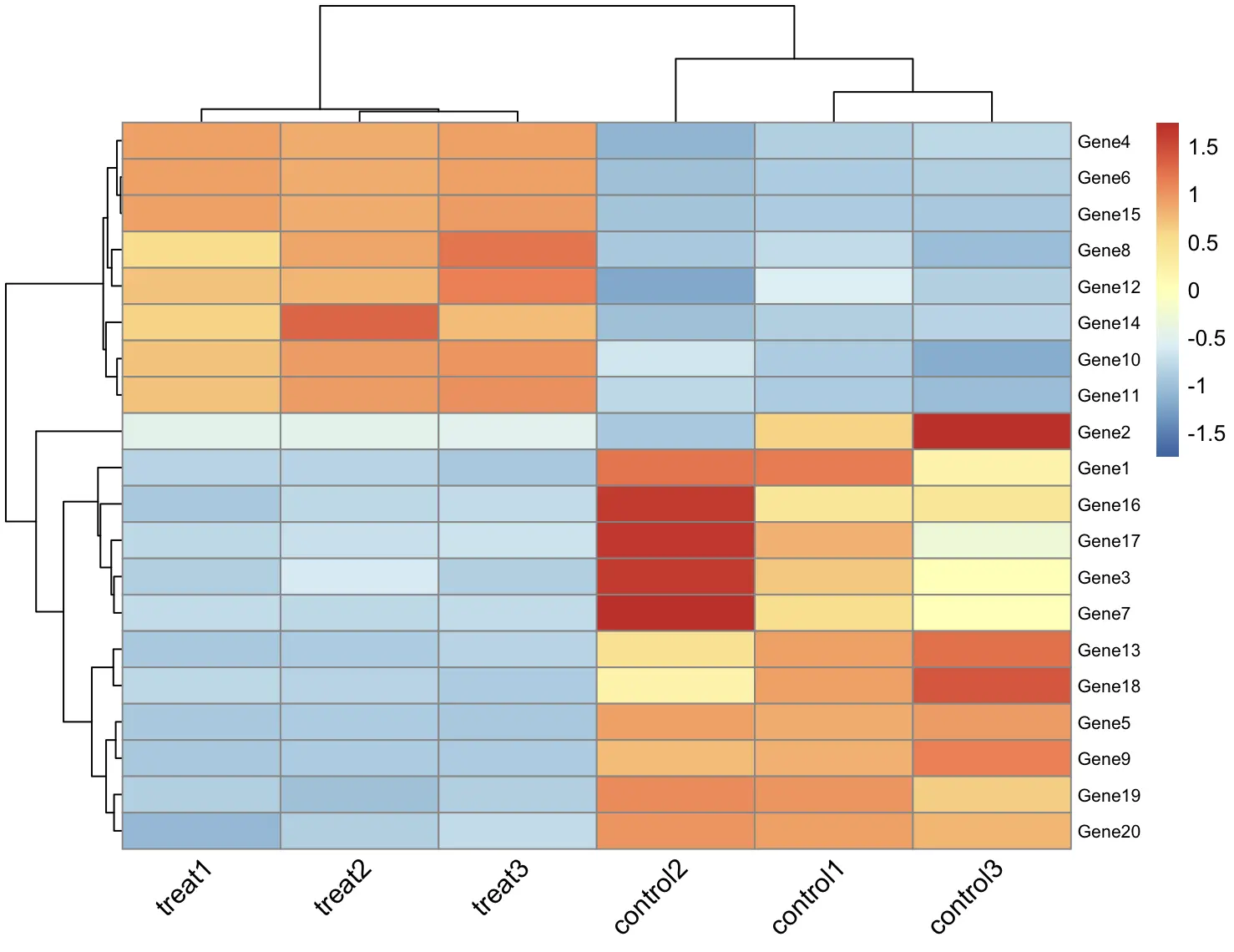
也可以使用 fontsize 参数统一行列标签的大小
也可以自定义行列标签
pheatmap(
df, scale = "row",
labels_row = paste0("Gene", LETTERS[1:20]),
labels_col = rep(c("Cancer", "Noraml"), each = 3)
)
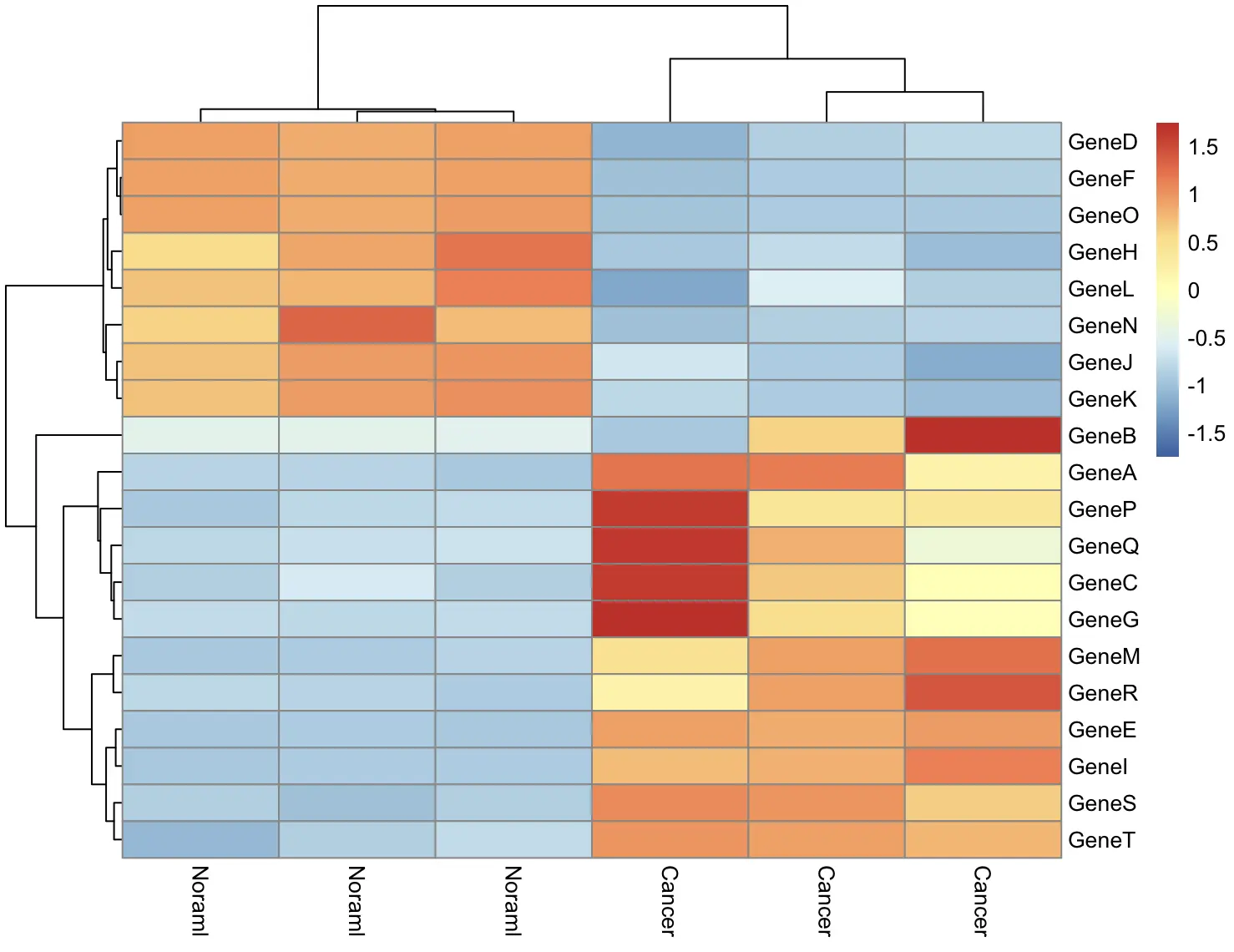
9. 注释
我们可以分别为行和列构建分组信息,例如对于行是基因,可以将其分为癌基因和抑癌基因等,而列为样本可以分为癌症和配对正常样本,同时样本对应的患者应该会有年龄性别等信息
例如
# 行注释信息
annotation_row <- data.frame(
GeneClass = sample(c("oncogene", "tumor suppressor"), size = 20, replace = TRUE)
)
rownames(annotation_row) <- paste0("Gene", 1:20)
# 列注释信息
annotation_col <- data.frame(
type = rep(c("Noraml", "Cancer"), each = 3),
Age = sample(30:60, 6)
)
rownames(annotation_col) <- colnames(df)
我们可以将这些信息以颜色条的方式添加到图中
pheatmap(
df,
scale = "row",
color = rainbow(7),
annotation_row = annotation_row,
annotation_col = annotation_col
)
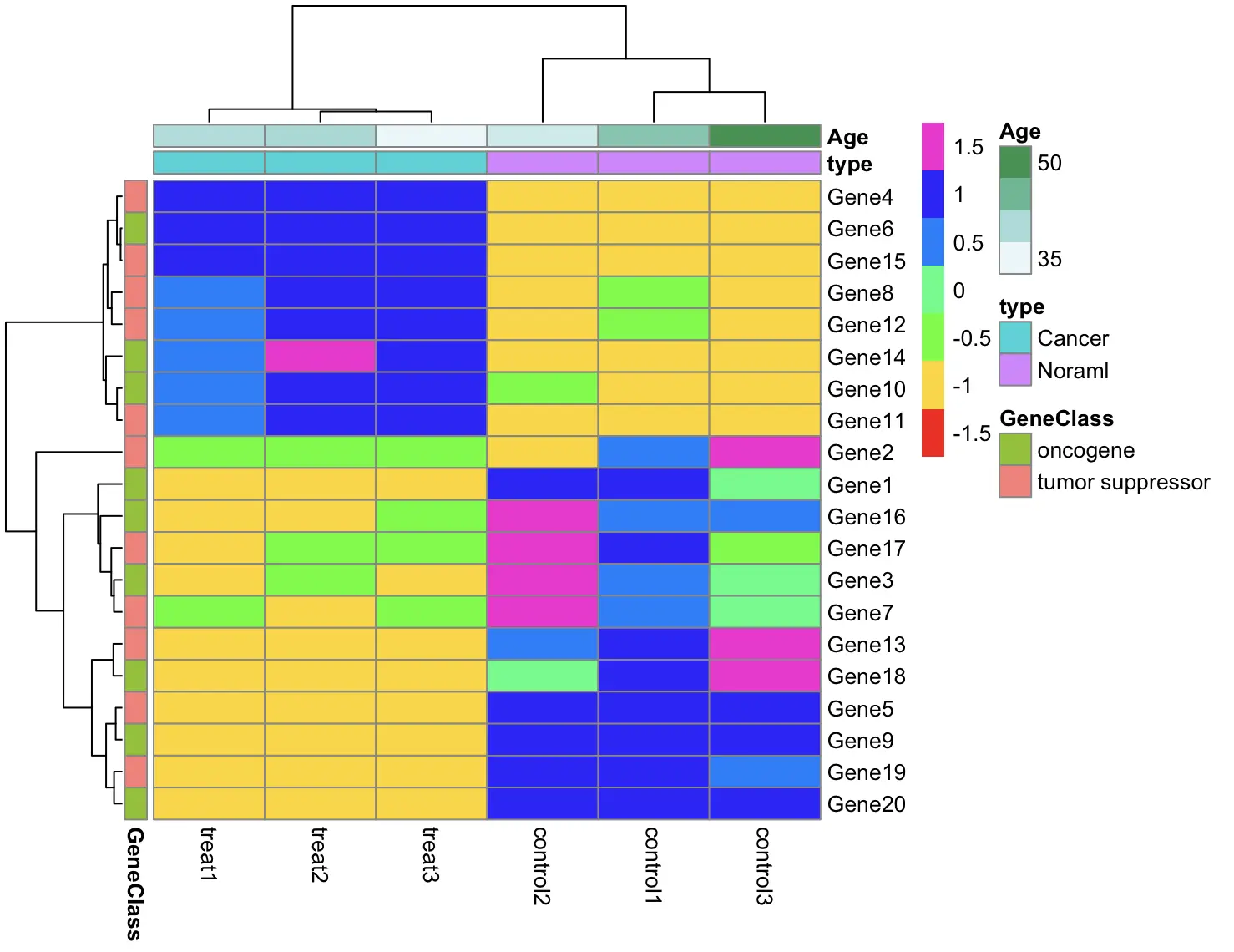
隐藏图例
pheatmap(
df,
scale = "row",
color = rainbow(7),
annotation_row = annotation_row,
annotation_col = annotation_col,
legend = FALSE,
angle_col = 45,
)
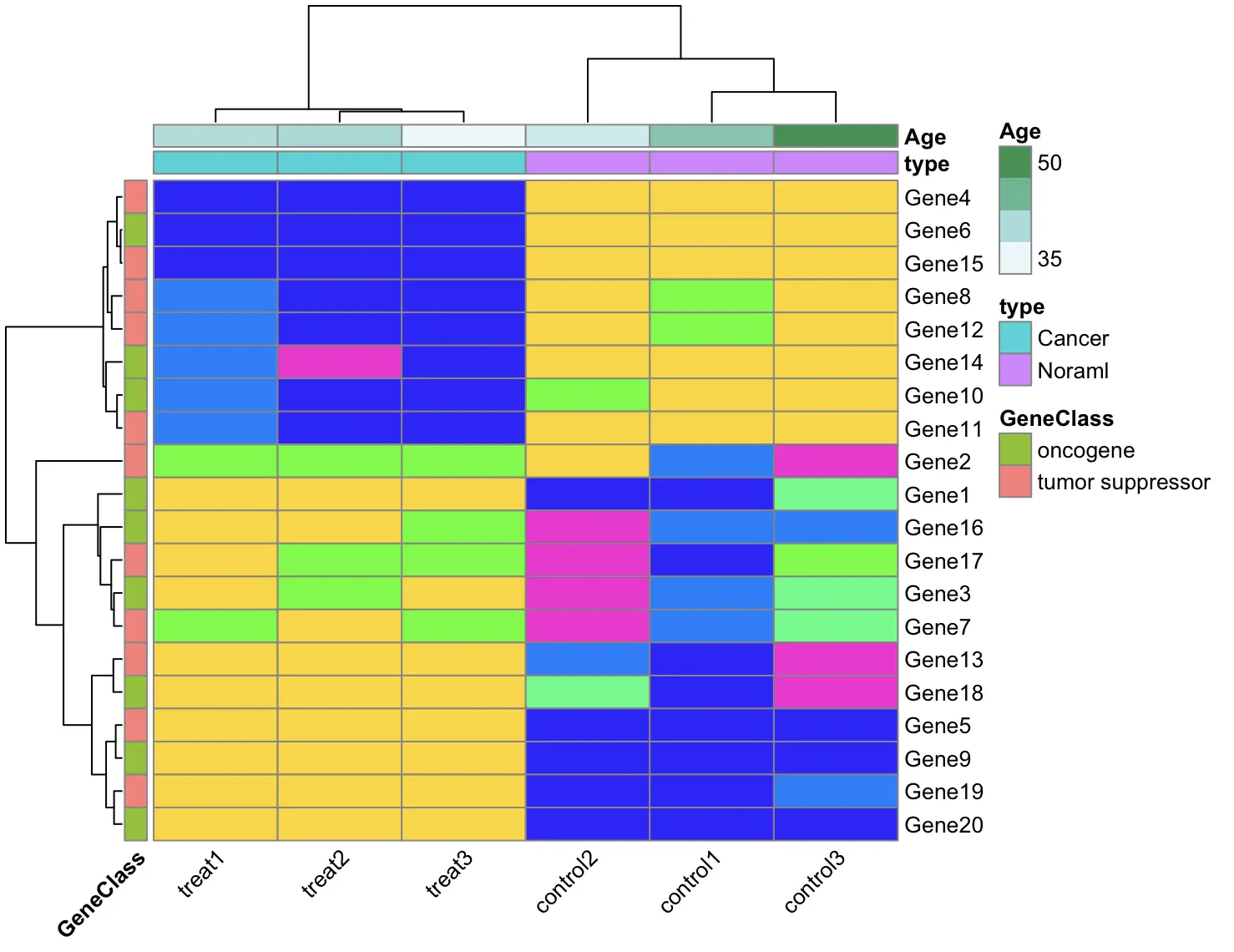
10. 对象信息
我们可以回去 pheatmap 函数返回的对象的信息
> p <- pheatmap(df, scale = "row")
> summary(p)
Length Class Mode
tree_row 7 hclust list
tree_col 7 hclust list
kmeans 1 -none- logical
gtable 6 gtable list
可以看到,返回对象 p 中包含 4 个变量,我们可以根据 tree_row 和 tree_col 提取出对应的行列顺序
> p$tree_row$order
[1] 4 6 15 8 12 14 10 11 2 1 16 17 3 7 13 18 5 9 19 20
> p$tree_col$order
[1] 4 5 6 2 1 3
提取这些信息有助于我们对数据进行分组,用于后续分析
参数列表

数据:
https://github.com/dxsbiocc/learn/blob/main/data/RPKM_DEG.csv