文章目录
1. Gromacs介绍
GROMACS是一个复杂的分子动力学模拟软件,用于模拟生物大分子的物理运动。
2. Gromacs安装
实机操作:Ubuntu20.04系统(Ubuntu 20.04.4 LTS版本)
安装Gromacs-2024 GPU-CUDA加速版
一、基础软件
安装gromacs提前要有 gcc , g++ , python , cmake ,Gromacs
检查方法
软件名 -version
软件名 -V
以下安装均须要管理员权限,本人一直在root身份运行,若需要 以下命令前可加上sudo
1. gcc下载安装
apt-get install gcc
2.g++下载安装
apt-get install g++
3.python
Ubuntu系统自带python3.8.10,没有的可以安装一下
apt install python
4.Cmake
apt-get install cmake
二、显卡驱动和CUDA安装
1.显卡驱动
Ubuntu20.04安装之后已经有显卡驱动,但版本较旧,需要更改。
在左下角显示应用程序中 选择 “软件与更新”
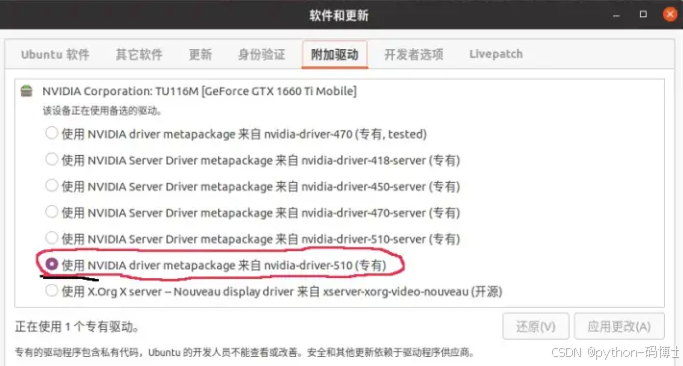
“附加驱动” 更改为合适的驱动版本,这里选择了510版本。
另可以 下载显卡驱动手动安装
参考:
https://blog.csdn.net/wf19930209/article/details/81877822
2. CUDA安装
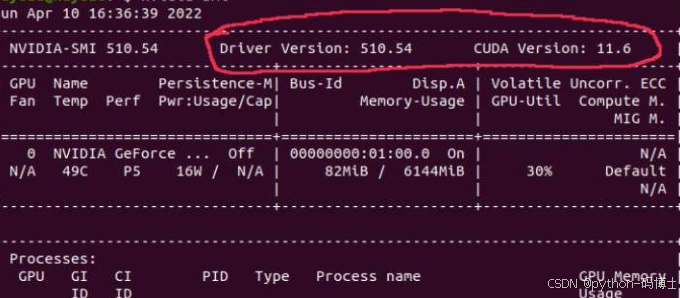
首先查看驱动适用CUDA版本
nvidia-smi
去Nvidia官网下载cuda安装包:https://developer.nvidia.com/cuda-toolkit-archive
本人选择deb安装方式
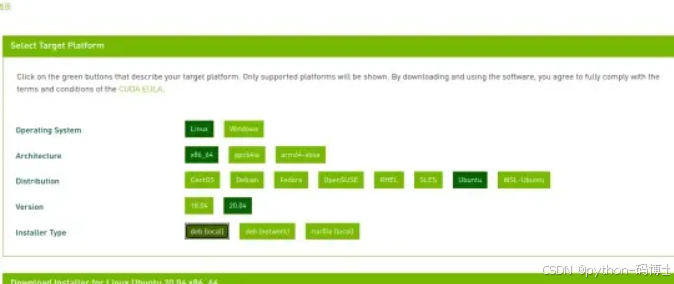
依次输入命令即可
配置环境变量
输入命令打开文件
vim ~/.bashrc
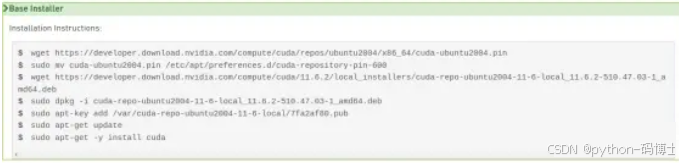
在文件最后输入以下语句
export PATH=/usr/local/cuda-11.6/binKaTeX parse error: Expected '}', got 'EOF' at end of input: {PATH:+:{PATH}}
export LD_LIBRARY_PATH=/usr/local/cuda-11.6/lib64KaTeX parse error: Expected '}', got 'EOF' at end of input: …LIBRARY_PATH:+:{LD_LIBRARY_PATH}}
(以上路径以各自安装的CUDA版本和安装路径 自行修改)保存
输入以下命令,更新环境变量配置
source ~/.bashrc
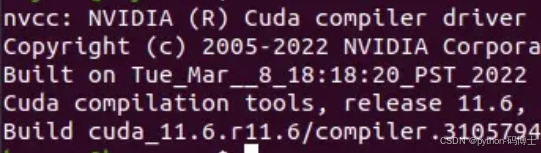
至此CUDA安装完成,输入nvcc -V命令查看CUDA信息
3.Gromacs-2024 GPU-CUDA安装
打开gromacs官网,下载最新版gromacs-2024
https://manual.gromacs.org/documentation/
也可以像我一样通过命令下载
wget https://ftp.gromacs.org/gromacs/gromacs-2024.3.tar.gz
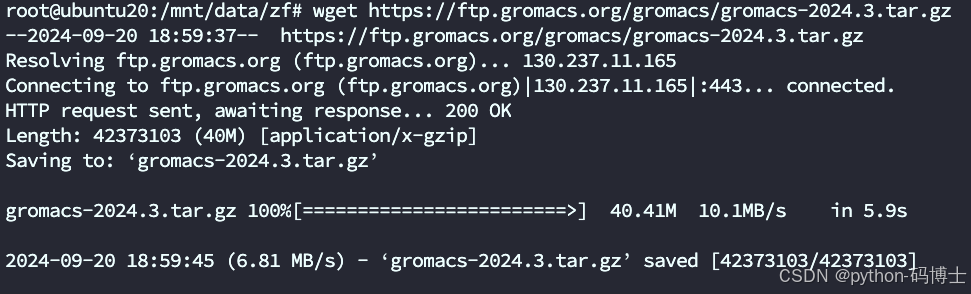
将tar.gz文件解压
tar xfz gromacs-2024.3.tar.gz
进入解压后的文件夹
cd gromacs-2024.3
mkdir build
cd build
在build目录下用cmake进行安装
cmake .. -DGMX_MPI=ON \
-DGMX_BUILD_OWN_FFTW=ON \
-DGMX_GPU=CUDA \
-DCUDA_TOOLKIT_ROOT_DIR=/usr/local/cuda \
-DCUDA_INCLUDE_DIRS=/usr/local/cuda/include \
-DCUDA_CUDART_LIBRARY=/usr/local/cuda/lib64 \
-DCMAKE_INSTALL_PREFIX=/mnt/data/zf/gromacs-2024.3
#(以实际CUDA tookit安装路径及版本为准)
make -j4 # 使用4个线程来编译,根据系统的 CPU 核心数调整这个参数以提高编译速度
make check
sudo make install
source /mnt/data/zf/gromacs-2024.3/bin/GMXRC
安装完成后输入
gmx -version
查看版本信息
若关闭后无法使用此命令 则可能是环境变量没有设置
设置环境变量
vim ~/.bashrc
在文件最后输入以下语句
source /mnt/data/zf/gromacs-2024.3/bin/GMXRC
(以上路径为 各自安装的Gromacs路径 自行修改)保存

输入以下命令,更新环境变量配置
source ~/.bashrc
重启终端后就可以输入
gmx --version
查看版本信息
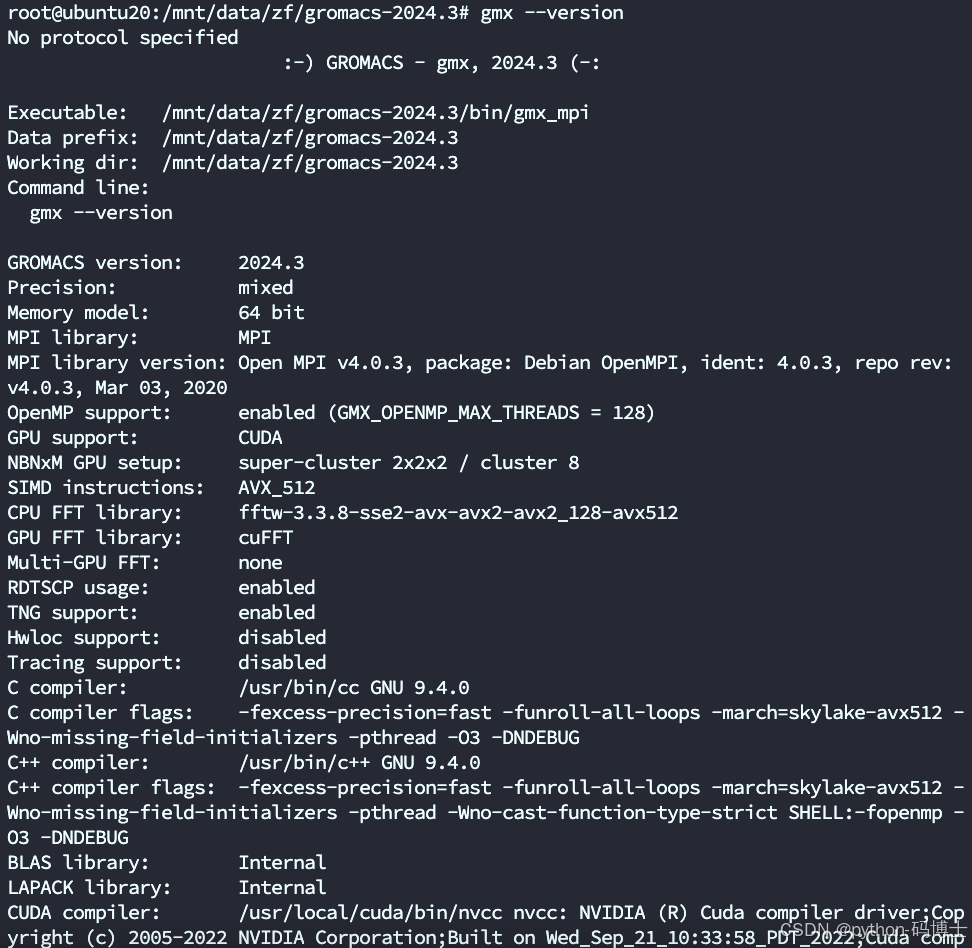
安装Gromacs-gpu-CUDA完成
本人比较过CPU与 GPU加速的运算速度
以Justin1 中 1AKI.pdb为例
NVT NPT 预平衡
仅使用CPU(I7-12700H)运算 :2.10h 2.00h
使用GPU加速(NVIDIA-RTX-3060)运算 :1min 1min
可能遇到的问题
1. 错误一
CMake Error at CMakeLists.txt:34 (cmake_minimum_required):
CMake 3.18.4 or higher is required. You are running version 3.16.3
-- Configuring incomplete, errors occurred!
原因:
CMAKE版本太低
解决方法:
- 卸载旧版本CMake:
sudo apt remove cmake
- 添加Kitware的APT仓库并安装最新版本:
sudo apt update
sudo apt install -y software-properties-common lsb-release wget
wget -O - https://apt.kitware.com/keys/kitware-archive-latest.asc | sudo apt-key add -
sudo apt-add-repository 'deb https://apt.kitware.com/ubuntu/ focal main'
sudo apt update
sudo apt install cmake
2.错误二
-- Looking for memalign
-- Looking for memalign - not found
-- MPI is not compatible with thread-MPI. Disabling thread-MPI.
-- Checking for module 'mpi-cxx'
-- No package 'mpi-cxx' found
-- Could NOT find MPI_CXX (missing: MPI_CXX_LIB_NAMES MPI_CXX_HEADER_DIR MPI_CXX_WORKS)
-- Could NOT find MPI (missing: MPI_CXX_FOUND CXX)
CMake Error at cmake/gmxManageMPI.cmake:87 (message):
MPI support requested, but no suitable MPI compiler found. Either set the
MPI_CXX_COMPILER to the MPI compiler wrapper (often called mpicxx or
mpic++), set CMAKE_CXX_COMPILER to a default-MPI-enabled compiler, or set
the variables reported missing for MPI_CXX above.
Call Stack (most recent call first):
CMakeLists.txt:550 (include)
原因:
目配置中启用了MPI(消息传递接口)支持,但CMake无法找到合适的MPI编译器。具体来说,MPI_CXX_COMPILER没有正确设置,系统找不到mpicxx或mpic++等MPI编译器。
解决方法:
sudo apt update
sudo apt install -y openmpi-bin openmpi-common libopenmpi-dev
3. 错误三
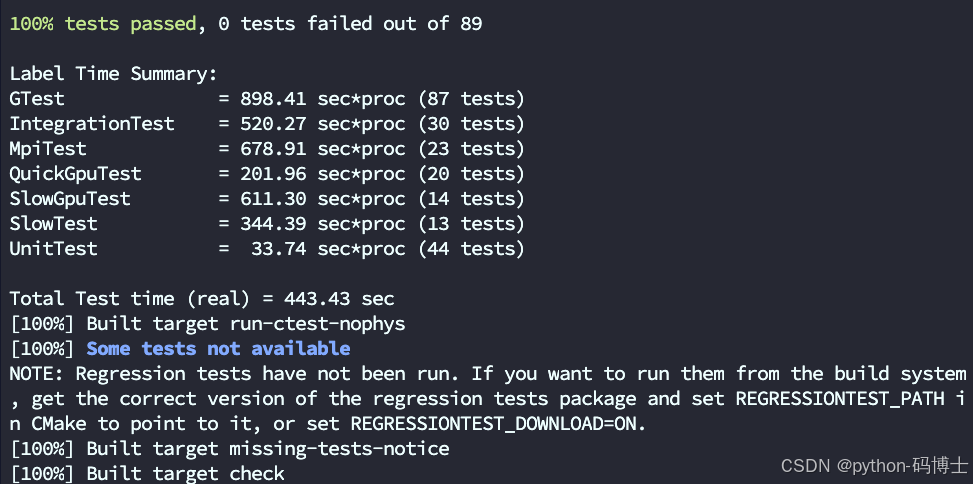
74% tests passed, 23 tests failed out of 89
Label Time Summary:
GTest = 221.64 sec*proc (87 tests)
IntegrationTest = 177.48 sec*proc (30 tests)
MpiTest = 0.58 sec*proc (23 tests)
QuickGpuTest = 53.97 sec*proc (20 tests)
SlowGpuTest = 124.45 sec*proc (14 tests)
SlowTest = 19.55 sec*proc (13 tests)
UnitTest = 24.60 sec*proc (44 tests)
Total Test time (real) = 135.57 sec
The following tests FAILED:
2 - GmxapiMpiTests (Failed)
4 - GmxapiInternalsMpiTests (Failed)
14 - TestUtilsMpiUnitTests (Failed)
16 - UtilityMpiUnitTests (Failed)
28 - DomDecMpiTests (Failed)
35 - MdrunUtilityMpiUnitTests (Failed)
66 - MdrunTestsOneRank (Failed)
67 - MdrunTestsTwoRanks (Failed)
69 - Minimize1RankTests (Failed)
70 - Minimize2RankTests (Failed)
73 - MdrunMpiTests (Failed)
74 - MdrunMultiSimTests (Failed)
75 - MdrunMultiSimReplexTests (Failed)
76 - MdrunMultiSimReplexEquivalenceTests (Failed)
77 - MdrunMpi1RankPmeTests (Failed)
78 - MdrunMpi2RankPmeTests (Failed)
79 - MdrunCoordinationBasicTests1Rank (Failed)
80 - MdrunCoordinationBasicTests2Ranks (Failed)
81 - MdrunCoordinationCouplingTests1Rank (Failed)
82 - MdrunCoordinationCouplingTests2Ranks (Failed)
83 - MdrunCoordinationConstraintsTests1Rank (Failed)
84 - MdrunCoordinationConstraintsTests2Ranks (Failed)
89 - MdrunVirtualSiteTests (Failed)
原因:MPI 执行失败的原因是由于你正在以 root 用户身份运行测试,mpiexec(MPI执行器)默认不允许以 root 身份运行,出于安全考虑这是 Open MPI 的默认行为。
解决方法:
- 以非 root 用户运行
如果你是 root 用户,先创建或切换到普通用户
su - your_non_root_user
- 允许 root 用户运行 MPI
如果你确实需要以 root 用户身份运行(不推荐,但如果你无法使用非 root 用户),你可以通过以下方式绕过限制。
export OMPI_ALLOW_RUN_AS_ROOT=1 # 表示你想允许以 root 用户运行。
export OMPI_ALLOW_RUN_AS_ROOT_CONFIRM=1 # 确认你已经了解并愿意承担这样做的风险。
或者,在 mpiexec 命令中使用 --allow-run-as-root 选项:
mpiexec --allow-run-as-root ...
然后再make check 就可以了
4.错误四
root@ubuntu20:/mnt/data/zf/gromacs-2024.3# source /mnt/data/zf/gromacs-2024.3/bin/GMXRCroot@ubuntu20:/mnt/data/zf/gromacs-2024.3# gmx --version
Command 'gmx' not found, but can be installed with:
apt install gromacs
原因:没有gmx可执行文件
解决方法:
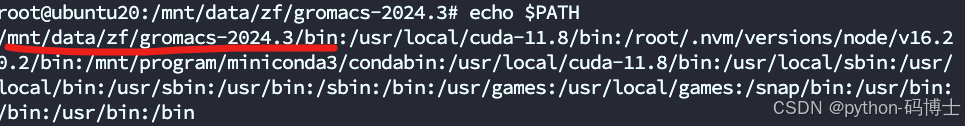
1.先用命令echo $PATH查看环境变量中是否存在gromacs的正确路径
2. 检查gromacs的bin目录是否有gmx
3.没有gmx但是有gmx_mpi,这意味着 GROMACS 已经正确安装并可以使用。gmx 可能是一个符号链接或别名,而不是一个单独的可执行文件。
如果你希望使用 gmx 而不是 gmx_mpi,可以创建一个符号链接。运行以下命令:

ln -s /mnt/data/zf/gromacs-2024.3/bin/gmx_mpi /mnt/data/zf/gromacs-2024.3/bin/gmx
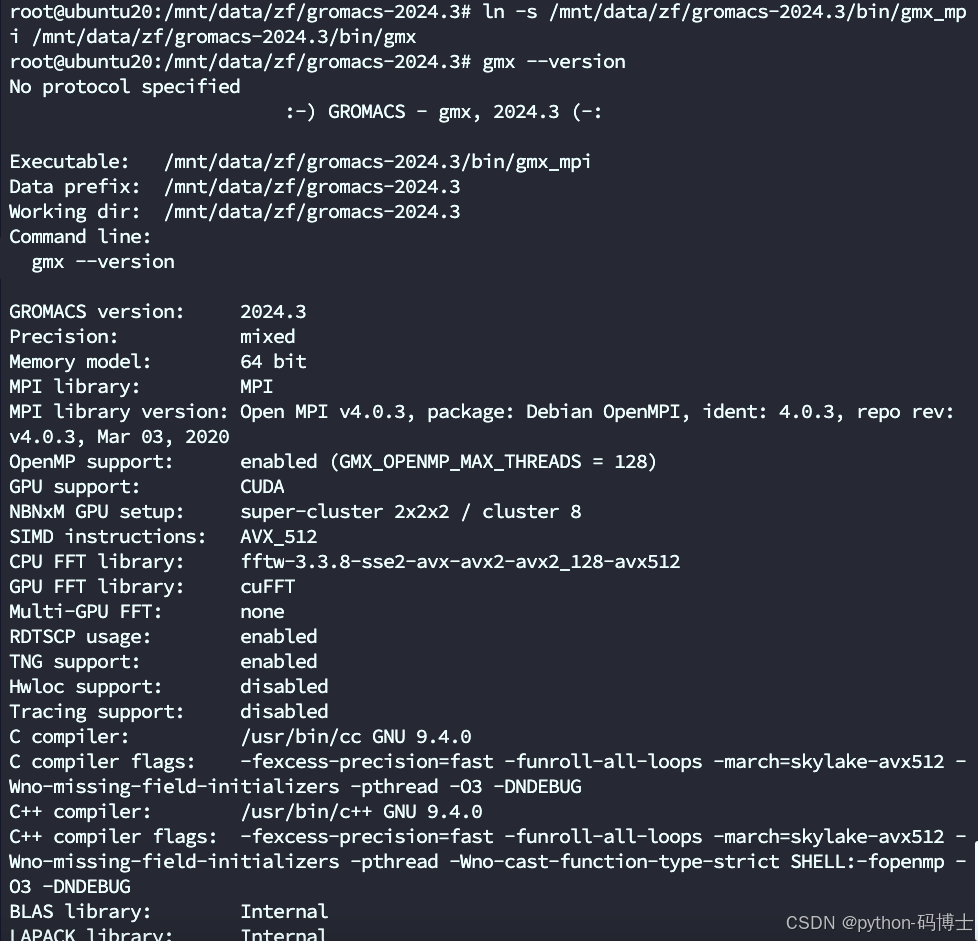
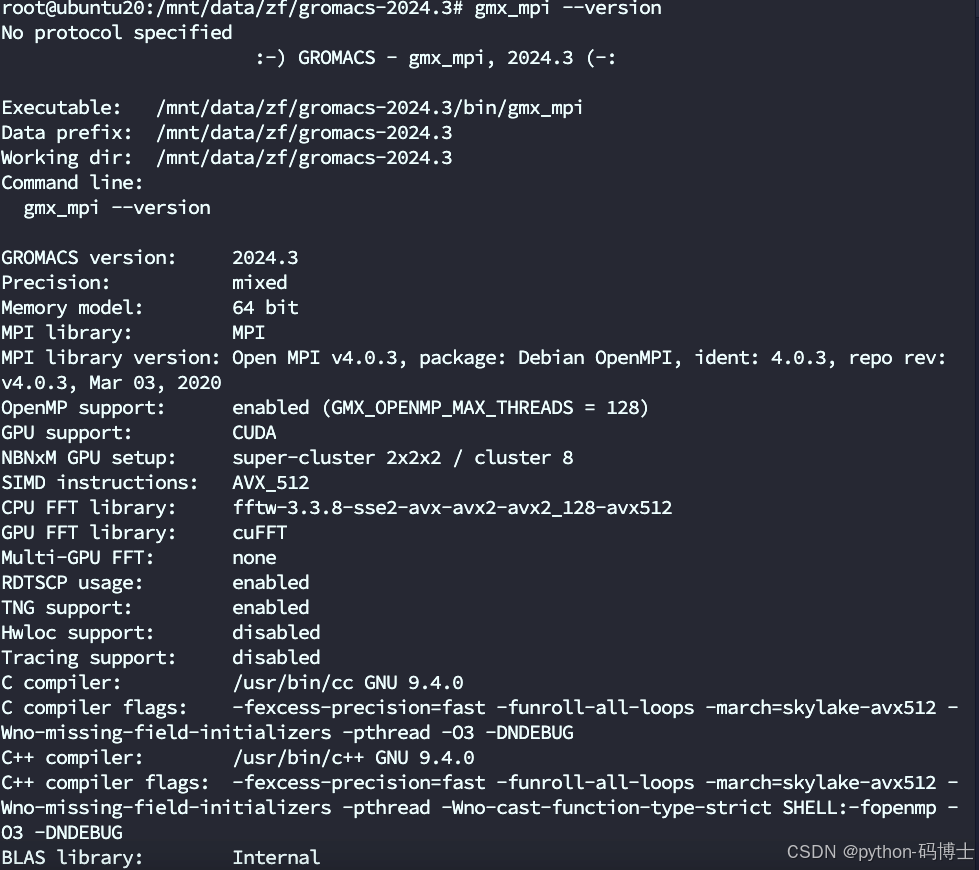
或者你忽略上面的链接步骤直接用 gmx_mpi --version命令
结束语
觉得不错的小伙伴,感谢点赞、关注加收藏哦!关注下方公众号获取更多学习资料!
