作者toby,来源公众号:Python生物信息学,python机器学习-乳腺癌细胞挖掘和生存分析(2024年新版)

随着人们生活水平提高,大家不仅关注如何生活,而且关注如何生活得更好。在这个背景下,精准治疗和预测诊断成为当今热门话题。
据权威医学资料统计,全球大约每13分钟就有一人死于乳腺癌,乳腺癌已成为威胁当代人健康的主要疾病之一,并且随着发病率的增加,死亡率也逐渐增加,作为女性实在不能不重视。
其中前十位死因;女性乳腺癌为首因,其余顺序与全人群死因一致。其中,乳腺癌可能没有任何先兆,是一个隐形杀手。
有不少人的乳腺癌是没有任何征兆的,有可能只是发现肿块但没有任何不舒服的地方,但一检查就确诊乳腺癌的情况不在少数,更甚至于有些人已经发展到乳腺癌晚期,只能采取切除患病乳房的方式来挽救生命。因此一定要密切注意乳房的变化,每年体检一次,排除患癌因素最保险。有权威数据统计,中国将进入乳腺癌高峰期,到2021年中国将有250万人患乳腺癌!保养胸部将不再是“喜不喜欢、需不需要”可选问题,在未来的日子里乳腺癌预防将是每个不同年龄女人的必须选择。
乳腺癌的主要症状包括:
1、肿块
为95%乳腺癌病人的乎发症状。大多单发,少见多发,形态偏于圆形、椭圆形或不规则形。质地一般较硬、境界不清。个别如髓样癌质地较软,境界较清。多发于外上象限,肿块增大较快,早期可有活动度。
2、皮肤改变
常见为浅表静脉怒张,酒窝征和桔皮样皮肤。炎性乳癌病人胸部皮肤可大片颜色变暗,呈硬结、增厚,杂以癌性斑块和溃疡呈铠甲状胸壁。晚期乳癌可向浅表溃破,形成溃疡或菜花状新生物。
3、乳头乳晕改变
乳房中央区乳腺癌,大导管受侵犯可致乳头扁平、凹陷、回缩,甚至乳头陷入晕下,导致乳晕变形。Paget氏病可出现乳头、乳晕皮肤湿疹样改变。
4、乳头溢液
乳腺癌伴溢液占乳癌总数的1.3-7%,且多见于管内癌、乳头状癌。血性溢液多见,次为浆液性、浆血性、水样等也有。以溢液为唯一症状乳癌,极少见,且大多为早期管内癌、乳头状癌,溢液乳腺癌多数先发现肿块后伴有溢液。
5、疼痛
早期出现的为无痛性肿块。乳癌合并囊性增生病时,可有胀痛、钝痛。晚期乳癌疼痛常提示肿瘤直接侵犯神经。
6、腋淋巴结肿大
作为乳腺癌首发症状少见(除非隐匿型乳腺癌)。大多提示乳腺癌病程进展,需排除上肢、肩背、胸部其他恶性肿瘤转移所致。

精准医疗和诊断预测离不开计算机编程,临床数据和机器学习算法。

乳腺癌是世界各地女性常见的癌症,通过尽早对患者进行临床治疗,尽早发现BC可大大改善预后和生存机会。因此,仅通过使用数据,python和机器学习就能帮助挽救生命真是太神奇了!通过下述代码,您已经完成创建乳房检测程序来预测患者是否患有癌症!同样,如果您愿意,您可以报名听我讲解课程的所有代码。
欢迎各位同学学习《python机器学习-乳腺癌细胞挖掘》课程2024年新版,教会大家建立诊断预测乳腺癌细胞模型
课程概述
此课程讲述如何运用python的sklearn快速建立机器学习模型。
课程有两个乳腺癌临床实战项目
1.(breast cancer Wisconsin)威斯康辛乳腺癌细胞挖掘和预测模型
2.(NCI SEER breast cancer)美国国立癌症研究所数据库乳腺癌生存分析和乳腺癌预测模型(2024年更新)

课程讲述十大经典机器学习算法:逻辑回归,支持向量,KNN,神经网络,随机森林,xgboost,lightGBM,catboost。这些算法模型可以应用于各个领域数据。课程涉及机器学习多个技术,包括stacking融合模型,非平衡数据处理,因子分析,pca主成分分析等等。
本视频系列通俗易懂,课程针对学生和科研机构,python爱好者。
本视频教程系列有完整python代码,观众看后可以下载实际操作。
了解癌症肿瘤基本常识,建立健康生活方式,预防癌症,减轻癌症治疗成本。
适用人群
研究生,博士生毕业论文,SEER/NCBI/SCI/Nature论文发布,python爱好者,机器学习,生物信息学,乳腺癌医学科研机构(课程有版权,引用需标注来源)
课程收益
0.癌症常识
1.python编程
2.机器学习十大经典算法建模
3.机器学习乳腺癌预测模型
4.机器学习乳腺癌生存分析
5.机器学习乳腺癌细胞预测模型
6.stacking融合模型技术
7.非平衡数据处理技术
8.因子分析和主成分分析技术

项目一.威斯康辛乳腺癌细胞挖掘的数据集
乳腺癌建模数据

项目二.2024年新项目基于美国国立癌症研究所数据库(NCI SEER)的乳腺癌生存分析和乳腺癌预测模型。

课程支持2个乳腺癌项目的预测模型数据和脚本下载。
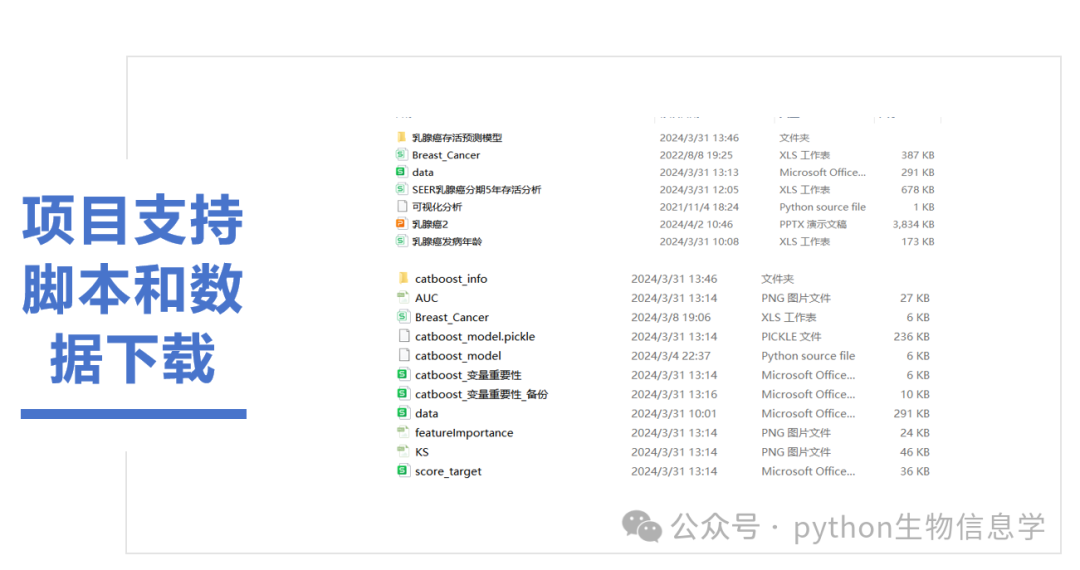

美国国立癌症研究所数据库(NCI SEER)的乳腺癌预测模型AUC大于0.86,模型性能优越。
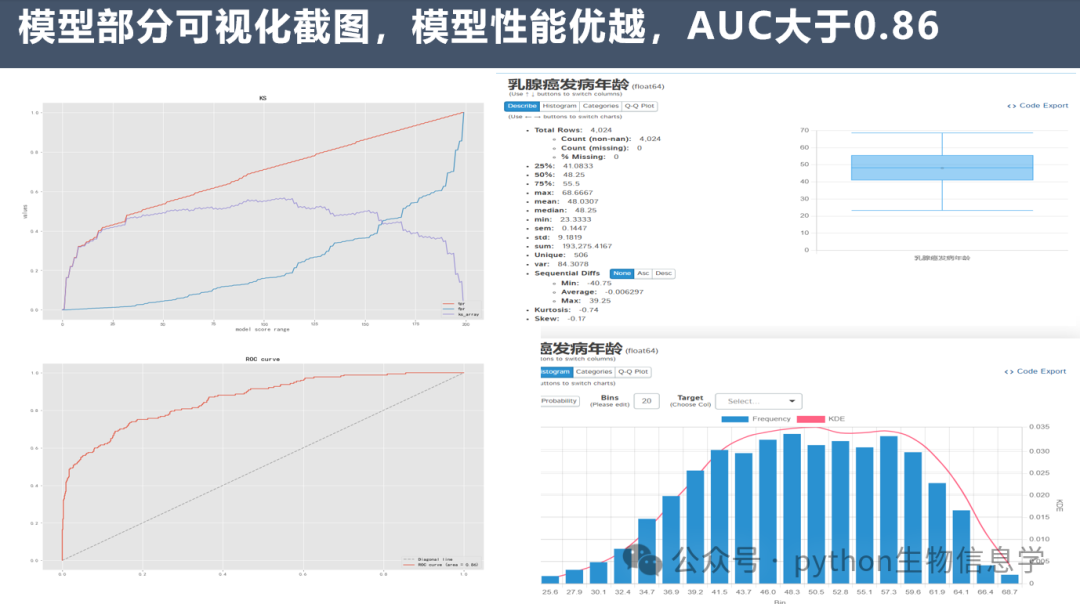
SEER数据库挖掘的文章正在逐年上升,并且发了不少高质量的文章。
课程中十大经典机器学习算法震撼登场:逻辑回归,支持向量,KNN,神经网络,随机森林,xgboost,lightGBM,catboost。课程提供视频里讲解脚本,这些模型脚本可以应用于各个领域数据,包括金融反欺诈模型,信用评分模型,收入预测模型等等,为中小企业提供现成解决方案。



随机森林变量权重可视化
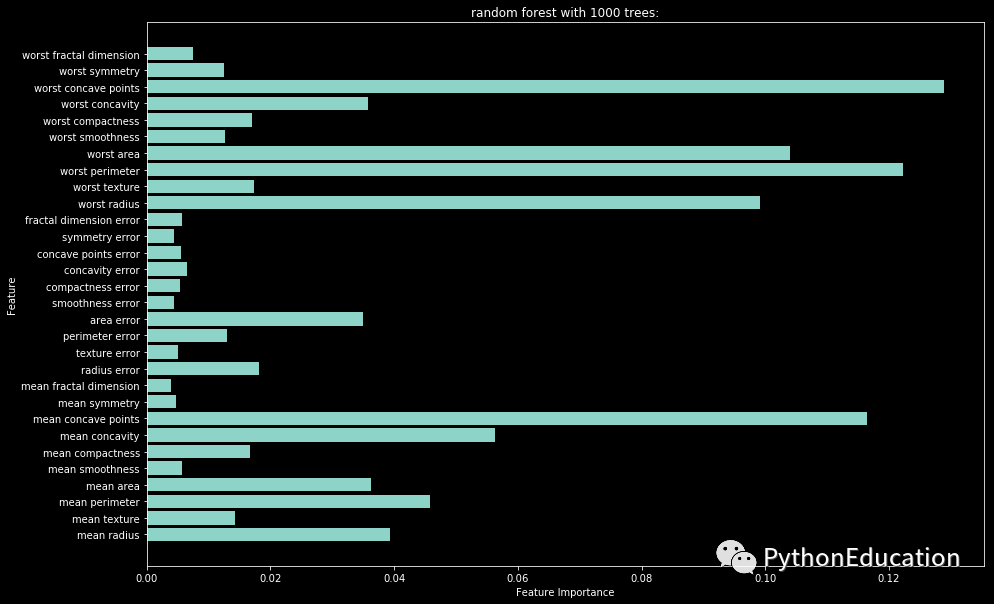
课程耗费三年时间,360度无死角的讲述整个模型开发周期,非市场上快餐教学。教程包括数据获取,数据预处理,变量筛选,模型筛选,模型评估,模型调参。
本视频系列通俗易懂,课程针对学生和科研机构,python爱好者。本视频教程系列有完整python代码,观众看后可以下载实际操作。这些模型代码可为中小型企业提供解决方案。

python机器学习编程环境搭建
python机器学习-乳腺癌细胞挖掘课程讲授初学者如何搭建python的Anaconda编程环境,Anaconda是一个集成数据科学编程框架,嵌入了sklearn,matplotlib,seaborn等常用机器学习和统计学包。
(1)下载anaconda
首先下载anaconda,这款框架比Python官网的编辑器更好用,下载网址为https://www.anaconda.com/download/
anaconda支持windows,linux,苹果操作系统
支持32位和64位操作系统
(2)导入sklearn第三方包
anaconda下载安装好后打开,自带sklearn第三方包
命令行输入import sklearn,无报错就表示运行正常
(3)pip install安装其他第三方包
机器学习中,有时候需要导入其他包,而sklearn没有,这时就需要用pip install安装其他第三方包

(4)非官方扩展包下载地址
有时候pip install安装失败,我们需要去欧文大学下载Python非官方扩展包
Python有大量非官方扩展包,应用于各行各业,主要是数据科学,人工智能,爬虫等等,下载地址为
https://www.lfd.uci.edu/~gohlke/pythonlibs/

乳腺癌细胞分类器建模
现在我们要用机器学习算法建立分类器,区分细胞为良性细胞或癌细胞。分类器就是解决二分类或多分类问题。
建立分类器算法很多,包括逻辑回归,xgboost,svm,神经网络等等。
开始编程:
在编写一行代码之前,我想做的第一件事是在代码的注释中加入描述。这样,我可以回顾我的代码并确切地知道它的作用。
#Description: This program detects breast cancer, based off of data.现在导入包/库,以使其更容易编写程序。
#import librariesimport numpy as npimport pandas as pdimport matplotlib.pyplot as pltimport seaborn as sns
接下来,我将加载数据,并打印数据的前7行。
注意:每行数据代表可能患有或未患有癌症的患者。
#Load数据#from google.colab 导入文件#用来加载数据在谷歌Colab #uploaded = files.upload() #使用在谷歌Colab负荷数据DF = pd.read_csv( 'data.csv')df.head (7)
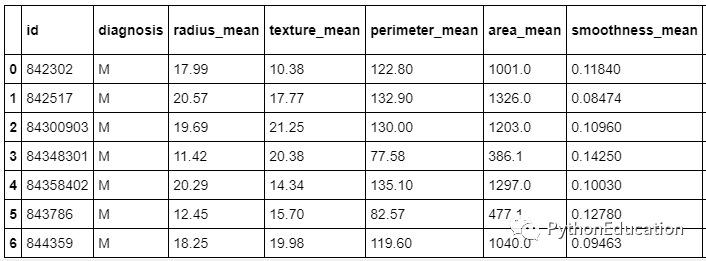
探索数据并计算数据集中的行数和列数。它们是569行数据,这意味着他们是该数据集中的569位患者,而33列则是每位患者的33个特征或数据点。
#计算数据集df.shape中的行数和列数
继续探索数据并获得包含空值(NaN,NAN,na)的所有列的计数。请注意,除了名为“ Unnamed:32 ”的列(其中包含569个空值)(数据集中的行数相同,这告诉我该列完全没有用)之外,所有列均未包含任何空值。
#计算每列df.isna()。sum()中的空值(NaN,NAN,na)

从原始数据集中删除“未命名:32 ”列,因为它没有任何值。
<em class="kq">#Drop the column with all missing values (na, NAN, NaN)</em><br><em class="kq">#NOTE: This drops the column Unnamed</em><br>df = df.dropna(axis=1)获取新的行和列数计数。
#Get the new count of the number of rows and colsdf.shape
获取具有恶性(M)癌细胞和良性(B)非癌细胞的患者数。
#Get a count of the number of 'M' & 'B' cellsdf['diagnosis'].value_counts()
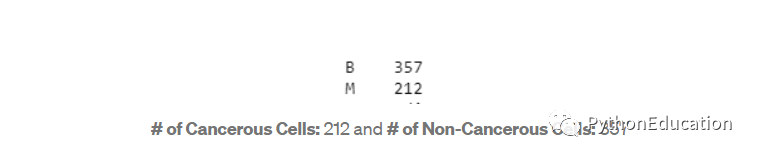
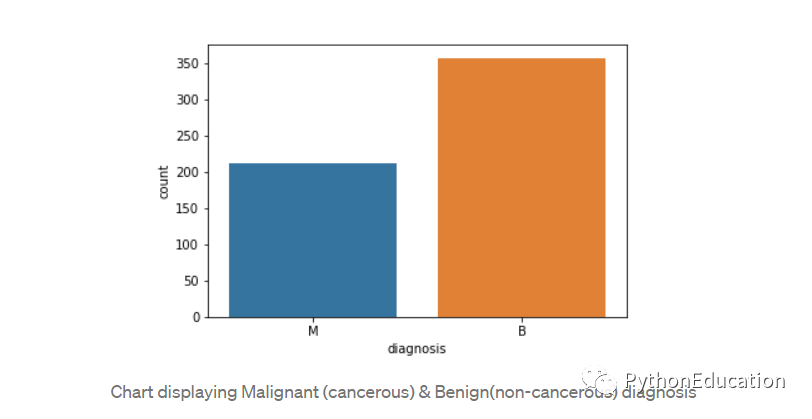
通过创建计数图可视化计数。
#Visualize this countsns.countplot(df['diagnosis'],label="Count")
查看数据类型以查看哪些列需要转换/编码。从数据类型中我可以看到,除“诊断”列外,所有列/功能都是数字,“诊断”列是在python中表示为对象的分类数据。
#Look at the data typesdf.dtypes

对分类数据进行编码。将“诊断”列中的值分别从M和B更改为1和0,然后打印结果。
#Encoding categorical data values (from sklearn.preprocessing import LabelEncoderlabelencoder_Y = LabelEncoder()df.iloc[:,1]= labelencoder_Y.fit_transform(df.iloc[:,1].values)print(labelencoder_Y.fit_transform(df.iloc[:,1].values))

创建一个对图。“对图”也称为散点图,其中同一数据行中的一个变量与另一变量的值匹配。
sns.pairplot(df,hue =“ diagnosis”)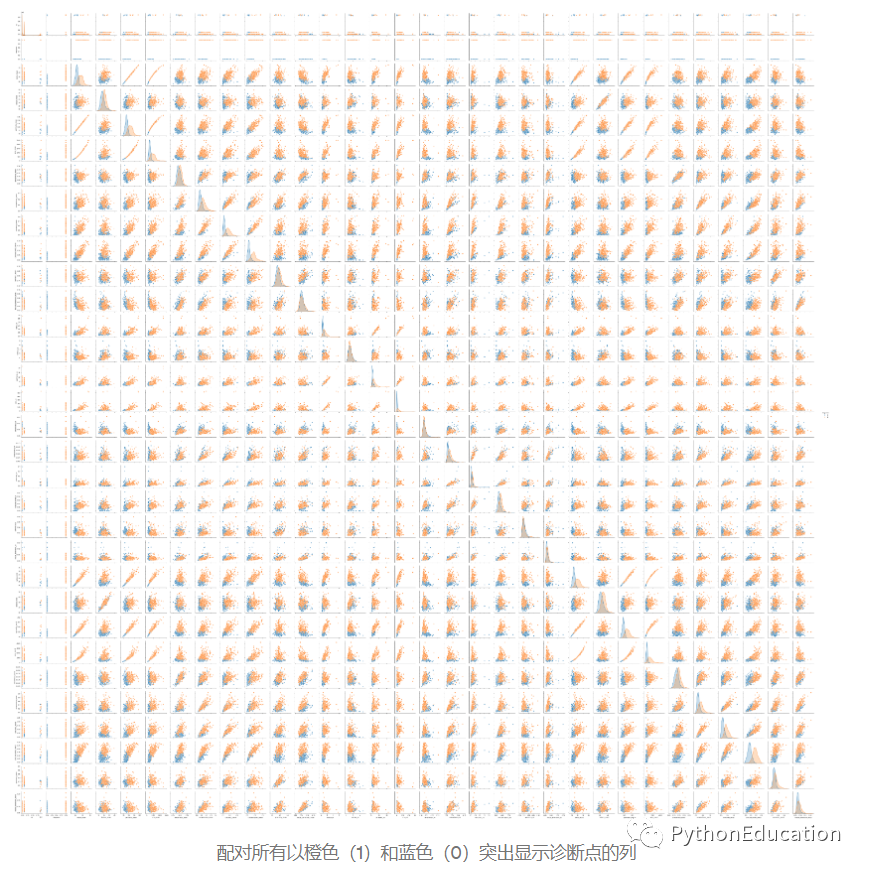
打印现在只有32列的新数据集。仅打印前5行。
df.head(5)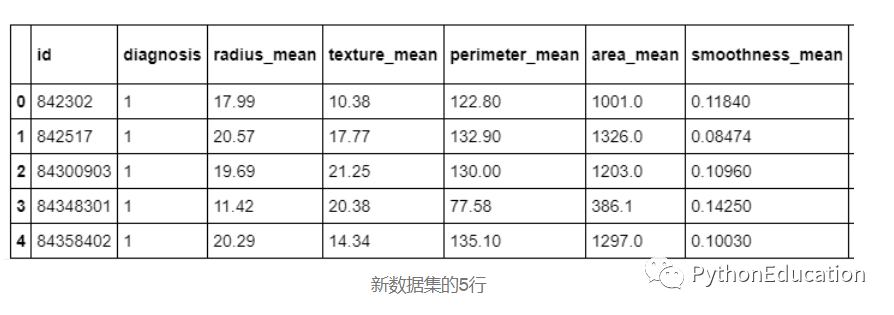
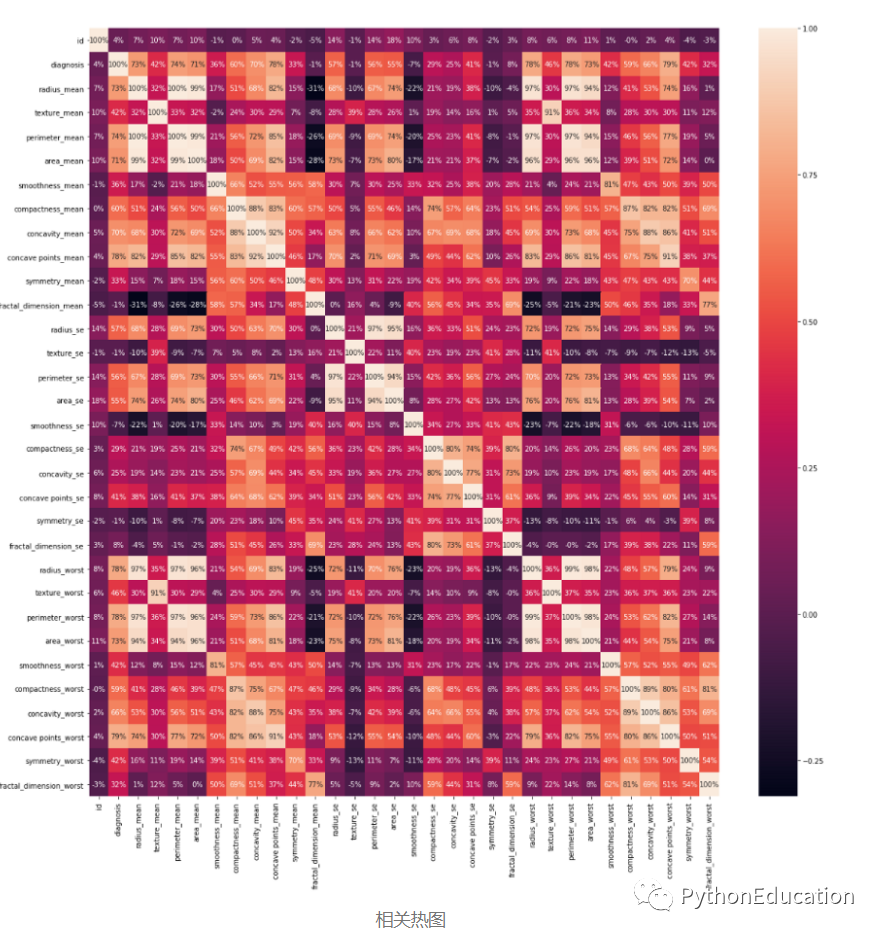
获取列的相关性。
#Get the correlation of the columnsdf.corr()

列相关样本
通过创建热图可视化相关性。
plt.figure(figsize =(20,20))sns.heatmap(df.corr(),annot = True,fmt ='。0%')
现在,我完成了探索和清理数据的工作。我将通过首先将数据集分为特征数据集(也称为独立数据集(X))和目标数据集(也称为从属数据集(Y))来设置模型的数据。
X = df.iloc [:, 2:31] .valuesY = df.iloc [:, 1] .values
再次拆分数据,但这一次分为75%的训练和25%的测试数据集。
from sklearn.model_selection import train_test_splitX_train,X_test,Y_train,Y_test = train_test_split(X,Y,test_size = 0.25,random_state = 0)
缩放数据以使所有要素达到相同的大小级别,这意味着要素/独立数据将处于特定范围内,例如0-100或0-1。
from sklearn.preprocessing import StandardScalersc = StandardScaler()X_train = sc.fit_transform(X_train)X_test = sc.transform(X_test)
创建一个函数以容纳许多不同的模型(例如,逻辑回归,决策树分类器,随机森林分类器)进行分类。这些模型将检测患者是否患有癌症。在此功能内,我还将在训练数据上打印每个模型的准确性。
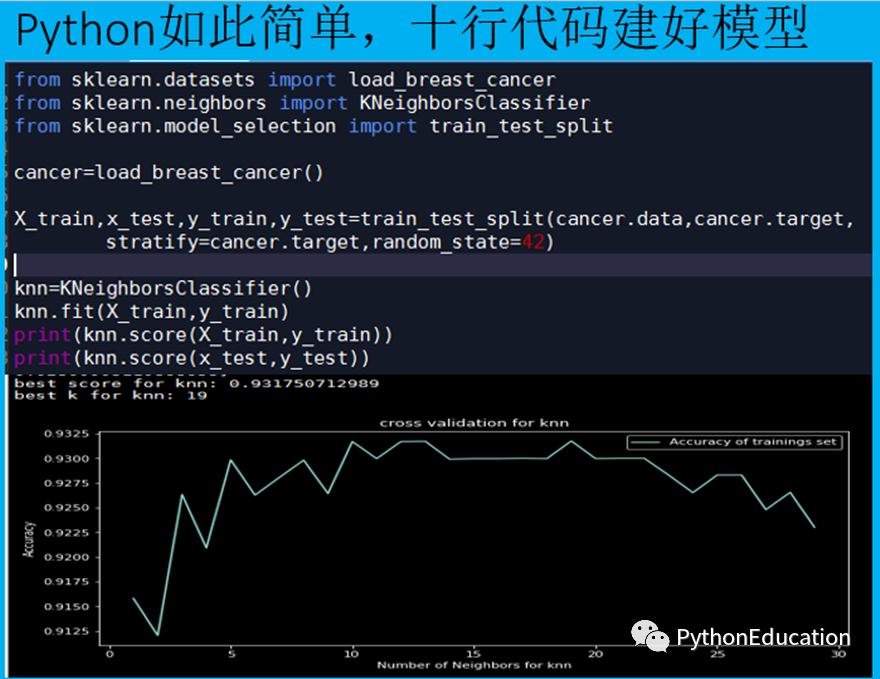
#学习链接:python机器学习-乳腺癌细胞挖掘 - 网易云课堂def models(X_train,Y_train):#Using Logistic Regressionfrom sklearn.linear_model import LogisticRegressionlog = LogisticRegression(random_state = 0)log.fit(X_train, Y_train)#Using KNeighborsClassifierfrom sklearn.neighbors import KNeighborsClassifierknn = KNeighborsClassifier(n_neighbors = 5, metric = 'minkowski', p = 2)knn.fit(X_train, Y_train)#Using SVC linearfrom sklearn.svm import SVCsvc_lin = SVC(kernel = 'linear', random_state = 0)svc_lin.fit(X_train, Y_train)#Using SVC rbffrom sklearn.svm import SVCsvc_rbf = SVC(kernel = 'rbf', random_state = 0)svc_rbf.fit(X_train, Y_train)#Using GaussianNBfrom sklearn.naive_bayes import GaussianNBgauss = GaussianNB()gauss.fit(X_train, Y_train)#Using DecisionTreeClassifierfrom sklearn.tree import DecisionTreeClassifiertree = DecisionTreeClassifier(criterion = 'entropy', random_state = 0)tree.fit(X_train, Y_train)#Using RandomForestClassifier method of ensemble class to use Random Forest Classification algorithmfrom sklearn.ensemble import RandomForestClassifierforest = RandomForestClassifier(n_estimators = 10, criterion = 'entropy', random_state = 0)forest.fit(X_train, Y_train)#print model accuracy on the training data.print('[0]Logistic Regression Training Accuracy:', log.score(X_train, Y_train))print('[1]K Nearest Neighbor Training Accuracy:', knn.score(X_train, Y_train))print('[2]Support Vector Machine (Linear Classifier) Training Accuracy:', svc_lin.score(X_train, Y_train))print('[3]Support Vector Machine (RBF Classifier) Training Accuracy:', svc_rbf.score(X_train, Y_train))print('[4]Gaussian Naive Bayes Training Accuracy:', gauss.score(X_train, Y_train))print('[5]Decision Tree Classifier Training Accuracy:', tree.score(X_train, Y_train))print('[6]Random Forest Classifier Training Accuracy:', forest.score(X_train, Y_train))return log, knn, svc_lin, svc_rbf, gauss, tree, forest
创建包含所有模型的模型,并查看每个模型的训练数据上的准确性得分,以对患者是否患有癌症进行分类。
model = models(X_train,Y_train)
catboost建模
今天我要介绍目前开源领域里最新的算法catboost。
catboost起源于俄罗斯搜索巨头yandex,准确率高,速度快,调参少,性价比高于xgboost
今天的CatBoost版本是第一个版本,以后将持续更新迭代。catboost三个特点:(1)“减少过度拟合”:这可以帮助你在训练计划中取得更好的成果。它基于一种构建模型的专有算法,这种算法与标准的梯度提升方案不同。(2)“类别特征支持”:这将改善你的训练结果,同时允许你使用非数字因素,“而不必预先处理数据,或花费时间和精力将其转化为数字。”(3)支持Python或R的API接口来使用CatBoost,包括公式分析和训练可视化工具。(4)有很多机器学习库的代码质量比较差,需要做大量的调优工作,”他说,“而CatBoost只需少量调试,就可以实现良好的性能。这是一个关键性的区别

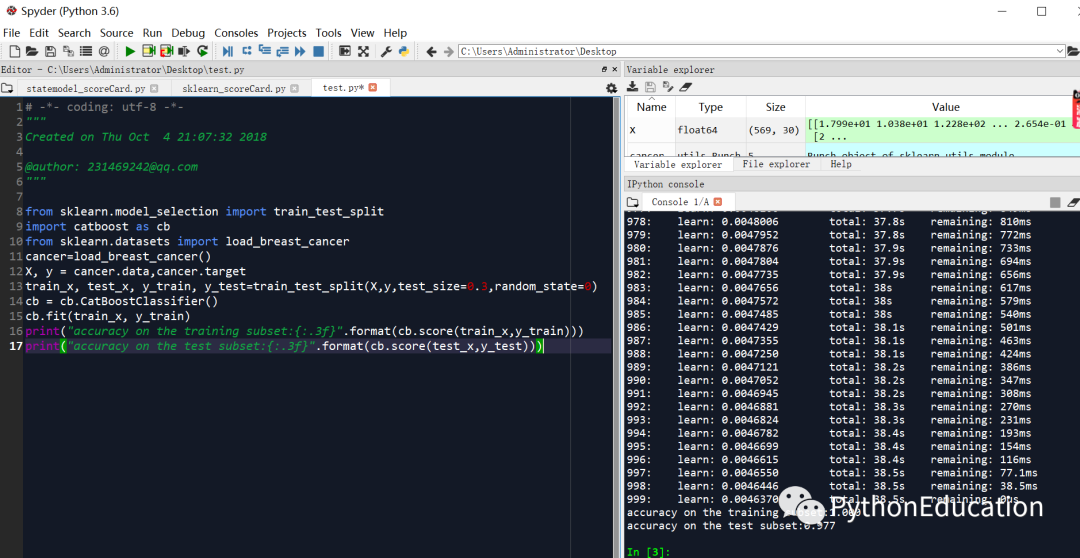
catboost建立乳腺癌分类器代码
# -*- coding: utf-8 -*-"""#学习链接:python机器学习-乳腺癌细胞挖掘 - 网易云课堂@author: [email protected]<br>微信公众号:python生物信息学"""from sklearn.model_selection import train_test_splitimport catboost as cbfrom sklearn.datasets import load_breast_cancercancer=load_breast_cancer()X, y = cancer.data,cancer.targettrain_x, test_x, y_train, y_test=train_test_split(X,y,test_size=0.3,random_state=0)cb = cb.CatBoostClassifier()cb.fit(train_x, y_train)print("accuracy on the training subset:{:.3f}".format(cb.score(train_x,y_train)))print("accuracy on the test subset:{:.3f}".format(cb.score(test_x,y_test)))
大家可以看到catboost预测准确率非常高,训练集100%,测试集97.7%
混淆矩阵
在测试数据上显示混淆矩阵和模型的准确性。该混淆矩阵告诉我们,每个模型有多少病人误诊(许多癌症患者是被误诊为不具有癌症又名假阴性,而谁没有癌症患者被误诊为患有癌症又名这个数字假阳性)和正确诊断的数量,真阳性和真阴性。
误报(FP) =测试结果错误地指示存在特定条件或属性。
真实阳性(TP) =灵敏度(在某些领域中也称为真实阳性率或检测概率)衡量正确鉴定出的真实阳性的比例。
真实阴性(TN) =特异性(也称为真实阴性率)衡量正确鉴定出的实际阴性的比例。
假阴性(FN) =测试结果,表明某个条件不成立,而实际上却成立。例如,测试结果表明某人实际患有癌症时没有罹患癌症

from sklearn.metrics import confusion_matrixfor i in range(len(model)):cm = confusion_matrix(Y_test, model[i].predict(X_test))TN = cm[0][0]TP = cm[1][1]FN = cm[1][0]FP = cm[0][1]print(cm)print('Model[{}] Testing Accuracy = "{}!"'.format(i, (TP + TN) / (TP + TN + FN + FP)))print()# Print a new line

其他获取模型指标的方法,以查看每个模型的性能如何。
#Show other ways to get the classification accuracy & other metricsfrom sklearn.metrics import classification_reportfrom sklearn.metrics import accuracy_scorefor i in range(len(model)):print('Model ',i)#Check precision, recall, f1-scoreprint( classification_report(Y_test, model[i].predict(X_test)) )#Another way to get the models accuracy on the test dataprint( accuracy_score(Y_test, model[i].predict(X_test)))print()#Print a new line

模型预测
测试数据中1–6个性能指标的模型样本
从以上的准确性和指标来看,在测试数据上表现最佳的模型是随机森林分类器,其准确性得分约为96.5%。因此,我将选择该模型来检测患者的癌细胞。对测试数据进行预测/分类,并显示“随机森林分类器”模型分类/预测以及显示或不显示他们患有癌症的患者的实际值。
我注意到了该模型,该模型将一些患者误诊为没有癌症而误诊为癌症,并且将确诊为癌症的患者误诊为未患癌症。尽管此模型很好,但在处理他人的生活时,我希望该模型更好,并使其准确性尽可能接近100%,或者至少好于医生。因此,有必要对每个模型进行一些调整。
#Print随机森林分类器模型的预测pred = model [6] .predict(X_test)print(pred)#打印空间print()#打印实际值print(Y_test)

Anaconda+KNN+网格调参+交叉验证

模型调参
python机器学习-乳腺癌细胞挖掘详细讲解模型调参技巧。调参是一门黑箱技术,需要经验丰富的机器学习工程师才能做到。幸运的是sklearn有调参的包,入门级学者也可尝试调参。
如果参数不多,可以手动写函数调参,如果参数太多可以用GridSearchCV调参,如果参数多的占用时间太长,可以用randomSizeCV调参,节约调参时间
GridSearchCV
如果参数太多可以用GridSearchCV调参
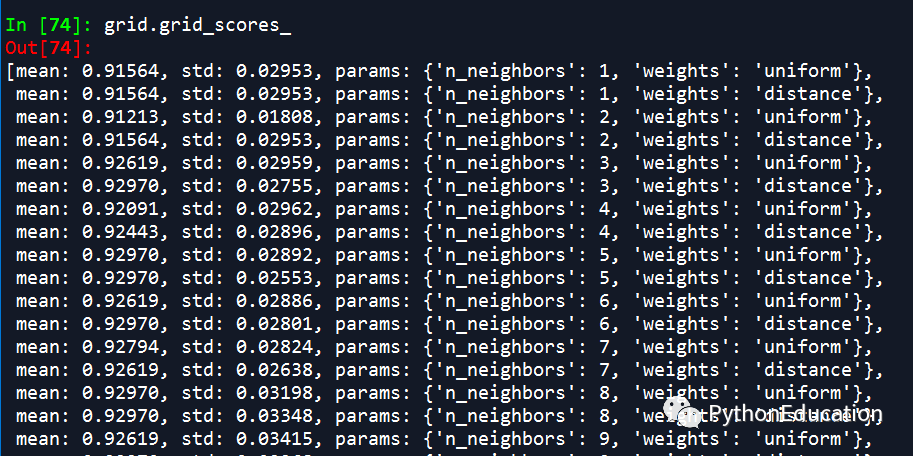
(1)单参数调参
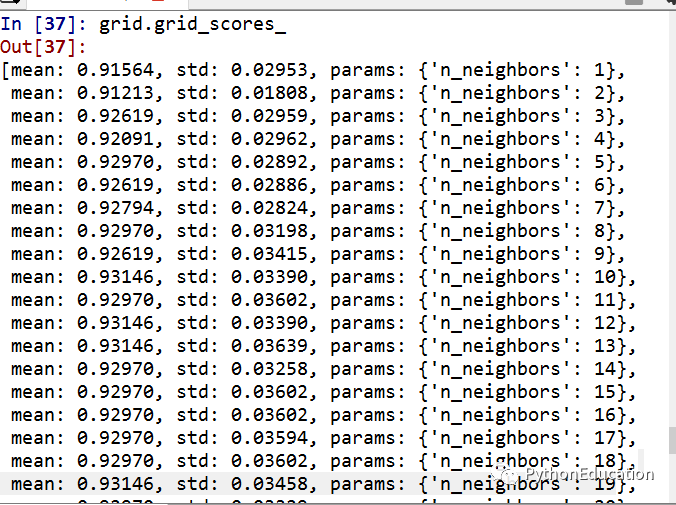
(2)多参数调参
因为有n_neighbors和weights两个参数,因此诞生了60个结果
因为有两个参数,所以得到最佳模型:weight=distance,n_neighbor=12
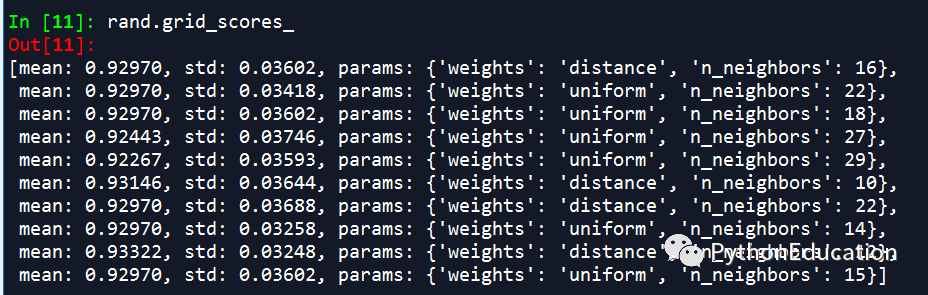
RandomSizeSearchCV
randomSizeCV调参类似于GridSearchCV的抽样
如果参数多的占用时间太长,可以用randomSizeCV调参,节约调参时间。
randomSizeCV调参准确率会略低于GridSearchCV,但可以节约大量时间。

randomSizeCV调参代码
# -*- coding: utf-8 -*-"""Created on Sat Jun 16 19:54:25 2018@author: [email protected]<br>微信公众号:pythonEducation"""from sklearn.grid_search import RandomizedSearchCVimport matplotlib.pyplot as plt#交叉验证from sklearn.cross_validation import cross_val_scorefrom sklearn.datasets import load_breast_cancerfrom sklearn.neighbors import KNeighborsClassifier#导入数据cancer=load_breast_cancer()x=cancer.datay=cancer.target#调参knn的邻近指数nk_range=list(range(1,31))weight_options=['uniform','distance']param_dist=dict(n_neighbors=k_range,weights=weight_options)knn=KNeighborsClassifier()#n_iter为随机生成个数rand=RandomizedSearchCV(knn,param_dist,cv=10,scoring='accuracy',n_iter=10,random_state=5)rand.fit(x,y)rand.grid_scores_print('best score:',rand.best_score_)print('best params:',rand.best_params_)
课程通过stacking融合模型,提升模型性能。
课程对乳腺癌数据集30个变量,用因子分析和主成分分析降维后,模型性能没有显著下降,反而略有上升。模型降维后,数据量减少,内存减少,运行和预测更快,模型部署难度降低,模型部署验证难度降低,企业开发模型时间成本降低,可谓一石十鸟。
课程通过非平衡数据处理技术,提升模型性能。
如果您们对疾病科研,人工智能预测模型项目感兴趣,欢迎各大医疗机构,科研机构,生物医药企业,研究生博士生论文联系。
项目联系人:重庆未来之智信息技术咨询服务有限公司,Toby老师,