Advances in the Application of Single-Cell Transcriptomics in Plant Systems and Synthetic Biology
单细胞转录组学在植物系统和合成生物学中的应用进展

摘要
植物是由多种细胞类型组成的复杂系统,其结构呈现出分层的组织形式。为了理解复杂植物系统的分子基础,单细胞RNA测序(scRNA-seq)已成为揭示细胞水平基因表达模式高分辨率以及研究细胞类型异质性的重要工具。此外,对植物生物系统进行scRNA-seq分析具有巨大潜力,可生成新知识以指导植物生物系统设计和合成生物学的发展。合成生物学旨在通过基因组编辑、工程化或基于理性设计的基因组重写,从而对植物进行遗传或表观遗传修饰,以提高作物产量和质量,推动生物经济发展并增强环境可持续性。特别是,scRNA-seq研究生成的数据可以用于开发高精度的“构建–设计–测试–学习”能力,以最大限度提高工程化植物生物系统的目标性能,同时将非预期副作用降至最低。迄今为止,scRNA-seq已经在少数植物物种中得到验证,包括模式植物(如拟南芥)、农业作物(如水稻)和生物能源作物(如杨树属植物)。未来,随着技术的进一步进步,scRNA-seq的成本有望降低,从而加速这一新兴技术在植物中的应用。在本综述中,我们总结了植物scRNA-seq的当前技术进展,包括样本制备、测序和数据分析,以为不同类型植物样本选择适当的scRNA-seq方法提供指导。随后,我们强调了scRNA-seq在植物系统生物学和植物合成生物学研究中的多种应用。最后,我们讨论了scRNA-seq在植物应用中的挑战与机遇。
引言
植物是由多种组织、器官和细胞类型组成的多细胞真核生物,这些结构整合并协调以执行特定功能[1]。传统的整体RNA测序(RNA-seq)技术不足以揭示植物复杂性背后的细胞类型特异性分子机制[2,3]。为了解决这一限制,单细胞RNA测序(scRNA-seq)被开发为一种先进技术,用于揭示细胞群体中存在的转录组变异,并捕获细胞水平上转录本丰度的动态变化[4]。相比整体RNA-seq,这项新技术具有多个优势:(a) scRNA-seq能够在复杂组织或细胞群体中识别和表征不同的细胞类型和状态,提供对细胞多样性的详细理解,而这些在整体RNA-seq中可能无法观测到[5,6];(b) scRNA-seq能够识别在整体RNA-seq中可能难以察觉的稀有细胞类型或亚群,研究其独特的基因表达谱和功能特性[7,8];(c) scRNA-seq可以捕获单个细胞中基因表达的时间动态变化,使得研究发育过程、细胞对刺激的响应或疾病进程的分辨率更高[9–11];(d) scRNA-seq提供了一种相对无偏的方法来研究单细胞水平的基因表达,能够检测整体RNA-seq中由于群体平均效应而可能遗漏的稀有或意外的基因表达模式[12]。
近年来,scRNA-seq已被用于植物系统生物学研究,探索细胞动态和调控网络。例如,scRNA-seq被应用于拟南芥,揭示热冲击条件下的根发育模式[13];在玉米中,用于研究胚胎发育过程中的细胞命运决定[14];在水稻和小麦中,用于理解胁迫条件下叶片和根细胞中的基因表达[7,8,15];在黑杨中,用于深入理解木本植物木质部分化的分子机制[16]。此外,scRNA-seq正逐渐成为促进植物合成生物学研究的强大技术,包括:(a) 识别细胞类型特异性生物部件(如启动子),以及(b) 解码植物再生机制以克服植物转化的瓶颈。例如,从scRNA-seq数据中识别的细胞类型特异性启动子可以用于细胞类型特异性的基因组工程,从而最大限度地减少负面的多效性影响[17]。
植物scRNA-seq的技术进步正在快速推进,包括细胞分离、文库制备和测序技术的改进[11,18]。针对植物特有挑战(如坚硬的细胞壁)开发了新的方法[11]。此外,包括可解释人工智能(AI)方法在内的计算工具和算法也被优化,以处理scRNA-seq生成的大量复杂数据集[19]。然而,仍存在一些挑战,例如全面的植物细胞图谱有限、捕获稀有细胞类型的难度,以及由于RNA含量低可能导致的技术噪声[9–11]。
目前已有多篇出色的综述总结了scRNA-seq在植物中的应用[11,20,21]。在本综述中,我们更新了scRNA-seq的技术发展情况,并概述了scRNA-seq在植物系统生物学和合成生物学中的最新进展。同时,我们讨论了这一激动人心领域的机遇与挑战,以激发在这一快速发展的领域中的进一步创新,并提出如何加速scRNA-seq在植物系统和合成生物学研究中应用的展望。
单细胞转录组学方法
scRNA-seq技术可以分为两个组成部分:用于生成scRNA-seq数据的实验部分和与分析scRNA-seq数据相关的计算部分。图1展示了scRNA-seq的一般工作流程。在本节中,我们将更新实验部分的相关内容,包括样本制备、scRNA-seq文库构建和测序。随后,我们总结了scRNA-seq数据计算分析的技术进展,以及为植物建立的scRNA-seq数据库。
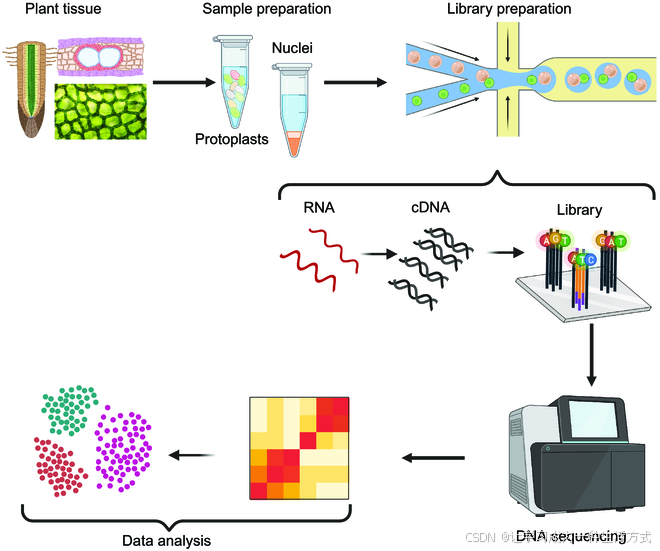
图1. 单细胞RNA测序(scRNA-seq)的概念性工作流程 从植物整体组织开始,解离细胞(原生质体)或从组织中提取细胞核,随后进行RNA提取、通过逆转录合成cDNA、文库制备、测序、表达丰度估算和细胞类型识别。本图重新绘制自[151],使用BioRender.com创建。
scRNA-seq的样本制备
在植物中,scRNA-seq实验已通过从不同物种的根、叶、茎尖分生组织、茎和花序中提取的原生质体和细胞核进行研究。使用细胞核样本的scRNA-seq也被称为单细胞核RNA测序(snRNA-seq)。尽管如此,大多数研究主要使用了原生质体,而非细胞核(见表1)。
原生质体因去除了细胞壁,能够捕获核RNA和胞质RNA。这种对完整转录组的全面捕获,可以为单个细胞内基因表达模式和调控过程提供更全面的视角。然而,用于scRNA-seq的原生质体制备存在一个主要局限性:酶解细胞壁会引发细胞应激,从而影响基因表达。
另一方面,细胞核的分离方法可以选择性捕获完整的细胞核,便于聚焦分析植物中核编码RNA转录本[22]。与基于原生质体的scRNA-seq方法相比,细胞核分离理论上能够分离包括对细胞降解酶敏感的复杂细胞类型,从而可能带来更高的细胞类型多样性[23]。
然而,使用细胞核样本进行scRNA-seq可能会错过来自胞质RNA的重要信息。这一限制影响了对转录后修饰、胞质信号传递及其他核外过程的研究能力,而这些过程对于全面理解细胞功能至关重要。
| Species | Sample preparation | Library construction | Sequencing platform | Reference |
|---|---|---|---|---|
| Arabidopsis thaliana | Root protoplast | Chromium Single Cell 3′ Reagent Kit v2 (10x Genomics) | Illumina NextSeq | [25] |
| A. thaliana | Root protoplast | Chromium Single Cell 3′ Reagent Kit v2 (10x Genomics) | Illumina HiSeq | [26] |
| A. thaliana | Root protoplast | Nextera XT DNA Library Kit (Illumina) | Illumina HiSeq and NextSeq | [32] |
| A. thaliana | Leaf protoplast | Chromium Single Cell 3′ Reagent Kit v2 and v3 (10x Genomics) | Illumina HiSeq | [30] |
| A. thaliana | Shoot protoplast | Chromium Single Cell 3′ Reagent Kits v3 (10x Genomics) | Illumina NovaSeq | [7] |
| Oryza sativa | Root protoplast | Chromium Single Cell 3′ Reagent Kit v3 (10x Genomics) | Illumina NovaSeq | [155] |
| O. sativa | Leaf protoplast | Chromium Single Cell 3′ Reagent Kit v2 (10x Genomics) | Illumina NovaSeq | [31] |
| O. sativa | Leaf and root protoplast | Chromium Single Cell 3′ Reagent Kit v3 (10x Genomics) | Illumina HiSeq | [15] |
| O. sativa | Inflorescence and leaf protoplast | The BD Rhapsody system | Illumina sequencer | [34] |
| Zea mays | Root protoplast | Chromium Single Cell 3′ Reagent Kit v3 (10x Genomics) | Illumina HiSeq | [156] |
| Solanum lycopersicum | Shoot apex protoplast | Chromium Single Cell 3′ Reagent Kit v3 (10x Genomics) | Illumina Novaseq | [137] |
| Populus spp. | Shoot apex and stem nuclei | Chromium Single Cell 3′ Reagent Kit v3.1 (10x Genomics) | Illumina NovaSeq | [22] |
表1. 植物scRNA-seq实验方法总结 表中总结了最近报告的植物scRNA-seq研究中样本制备、文库构建和测序平台的信息。
微流控平台的使用是单细胞分离、隔离和分析的主要工作流程[24]。为了确保最佳性能,原生质体或细胞核悬液必须由充分解离的完整原生质体/细胞核组成,且无任何细胞碎片。目前,与微流控兼容的原生质体分离方法的开发主要集中在拟南芥[7,25,26]。如上所述,原生质体分离的一个主要挑战是从坚硬的细胞壁中解离细胞。许多物种中的某些组织(例如木质部)对酶解具有较高的抗性[27,28]。
作为替代方案,可以使用荧光激活细胞分选(FACS)来分离和纯化细胞核,以应对酶解细胞壁带来的挑战。FACS还具有其他优势,例如可以使用冷冻样本和减少悬液制备时间[29]。在用于植物scRNA-seq的平台中,原生质体和细胞核都可以通过与不同组织类型兼容的10x Genomics协议轻松适配[15,22,25,26]。植物scRNA-seq中选择使用原生质体还是细胞核取决于具体的研究目标、植物物种以及转录组覆盖范围、空间信息和技术可行性之间的权衡。研究人员应仔细考虑这些因素以确定其研究中最适合的样本制备方法。
scRNA-seq文库构建与测序
在植物scRNA-seq研究中应用了多种文库制备方法,其中10x Genomics系统被广泛用于构建多种植物物种的scRNA-seq文库(见表1)。该系统可用于分析来自多种植物组织的5,000至20,000个细胞[25,26,30,31]。除了常用的10x Genomics系统外,其他文库构建方法也在植物scRNA-seq中展现了巨大潜力。例如,Nextera XT DNA文库制备试剂盒(Illumina)被用于构建scRNA-seq文库,分析了来自拟南芥根部的超过12,000个细胞[32]。最近,BD Rhapsody系统(BD Life Sciences, San Jose, USA)也被用于scRNA-seq研究。该系统利用基于微孔板的盒式设备捕获广泛范围的单细胞,同时测量多个方面,包括基因表达和蛋白质丰度[33]。使用该系统分析了来自水稻花序的37,571个细胞,从而生成了一个关于花序早期发育的综合基因表达图谱[34]。通过各种方法构建的scRNA-seq文库使用Illumina测序技术(如HiSeq、NextSeq和NovaSeq)进行测序(见表1)。
scRNA-seq数据分析
scRNA-seq实验会生成复杂的高维数据,这需要专门的流程进行分析和解读[35]。scRNA-seq数据分析包含多个步骤,包括预处理、比对(或映射)、质量控制、标准化、数据补全、降维、数据整合、聚类、差异基因表达分析以及功能分析[36–37],如图2所示。
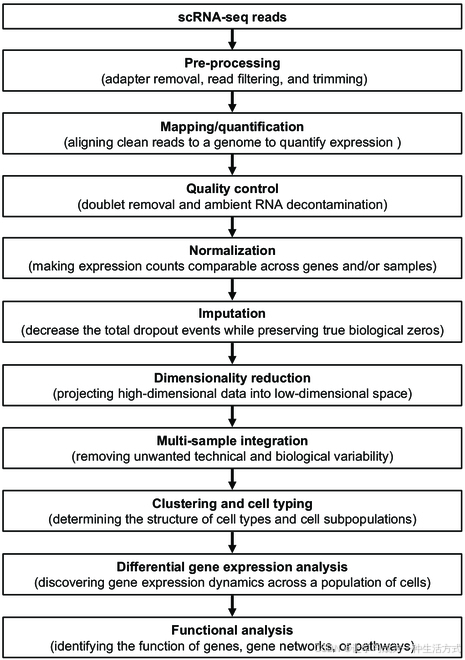
图2. scRNA-seq数据分析流程的示意图 图中展示了scRNA-seq数据分析的主要流程,重新绘制自[35–37]。
预处理
预处理步骤包括过滤低质量的读取(例如,具有低质量碱基的唯一分子标识符[UMI]读取)以及修剪低质量碱基[38,39]。
读取比对
最常用的scRNA-seq数据读取比对工具包括Cellranger [40]、STARsolo [37,41,42]和Kallisto [43]。Cellranger包含一个经过修改的STAR版本,与STARSolo功能相当,但效率较低。然而,还有许多其他可供选择的比对工具。不论选择哪种比对工具,读取数据都会比对到参考基因组,并通过UMI进行去重,生成一个基因与细胞的整数字特征矩阵。
质量控制
质量控制步骤包括去除双细胞/多细胞(由两个或更多细胞生成的文库)以及清除环境RNA(来自未标记裂解细胞的mRNA)污染[35]。双细胞和多细胞会严重干扰scRNA-seq数据分析,目前已有多种工具用于检测这些问题,例如DoubletFinder [44]和SoCube [37]。环境RNA污染是scRNA-seq的一个重要问题,尤其是在基于单细胞核分离的方法中,裂解细胞会导致环境RNA污染[45]。通常,通过以下几个阈值步骤省略大量比对的细胞:较低的UMI下限(<500 UMIs)、较低的独特基因下限(<50个基因),以及低线粒体含量下限(通常为5%到10%,具体取决于植物物种,且不适用于snRNA-seq)。
标准化
scRNA-seq数据的标准化通常分为两个步骤。首先,缩放步骤将每个细胞的特征计数除以该细胞的总计数,以解决基因特异性偏差(如相对基因丰度),然后对所有表达值按同一比例缩放。这一步类似于常见的整体RNA-seq缩放步骤(如每百万计数[CPM]或每百万转录本[TPM]),虽然scRNA-seq协议使用UMI,且不表现出整体RNA-seq方法中的基因长度偏差[46],因此无需考虑此问题。其次,所有特征计数应用一个转换(通常是对数转换),以实现方差稳定化[36,47,48]。样本间标准化可以通过两类方法完成:(a) 全局缩放方法,包括简单的CPM转换及更稳健的缩放程序,如DESeq2的标准化方法[49]和PsiNorm [50];(b) 非线性方法,如Linnorm [51]、sctransform [52]和Dino [53]。
数据补全
scRNA-seq数据高度稀疏,难以确定观察到的零值是生物学零值还是技术零值(即所谓的“掉落事件”)[54]。然而,已开发了多种方法来减少计数矩阵中的掉落事件,同时保留真实的生物学零值。这些补全方法分为三大类:
-
使用概率模型来对掉落事件的基因表达值进行补全。
-
对整个计数矩阵进行处理,通过平滑细胞中的基因表达值和识别相似的细胞谱系来实现。
-
基于深度学习或低秩矩阵方法,从细胞谱系的潜在空间表示重构整个计数矩阵[55]。
一项基准研究对18种不同的补全方法进行了比较,发现单细胞变分推断(scVI)[56]、深度计数自动编码网络(DCA)[57]和基于马尔可夫亲和图的细胞补全(MAGIC)在所有方法中排名较高[58]。此外,这项研究显示k近邻(kNN)平滑[59]和scVI [56]在scRNA-seq与整体RNA-seq之间差异表达基因(DEGs)的重叠率最高,相较于未补全的数据,显著提高了差异表达分析的表现[55]。此外,MAGIC [58]、SAUCIE [60]和SAVER-X [61]在提高无监督聚类结果方面排名最高,相较于未应用补全方法时表现更优[55]。
降维
降维步骤将高维数据投影到低维空间,以便可视化细胞簇结构和推断细胞发育轨迹[62]。多种降维方法已被开发,包括基于线性和非线性模型的方法以及深度学习算法。目前最常用的方法是统一流形近似与投影(UMAP),它能够为细胞类型簇提供出色的组织效果,并且计算成本最低[63]。最近,一些新的降维方法也被开发出来,例如DREAM,可捕获细胞类型间的差异,自动学习输入数据的层次表示,并对掉落事件建模[64];以及PHATE,它生成的低维嵌入比现有的可视化方法具有更好的去噪效果[65]。
数据整合
整合可以在样本之间或条件之间进行。多样本整合步骤旨在消除不需要的技术和生物学变异[35]。许多工具已被开发用于整合由scRNA-seq数据衍生的异质样本,例如批次平衡k近邻(BBKNN)[66]、Harmony [67]、Seurat CCA [68]和Scanorama [69],其中Scanorama的表现最佳[35]。
聚类与细胞类型划分
在scRNA-seq实验中,为了将细胞分配到某个细胞类型或状态,需要将细胞划分为具有相似基因表达模式的聚类。聚类包括两个步骤:从基因-细胞计数矩阵生成一个图,然后对该图进行聚类。一个广泛使用的图嵌入方法是k近邻方法(kNN)[70],不过也可以使用其他图生成方法,包括解释性人工智能方法,如iRF-LOOP和CoMet [71]。常用的图聚类方法有许多,其中包括k均值聚类、层次聚类[72]、Leiden算法[73]和Markov聚类[74]。一旦细胞根据基础图拓扑被划分到聚类中,每个聚类都会根据其中的主要差异表达基因(DEGs)分配一个细胞类型。
差异表达与功能分析
差异表达分析用于发现细胞群体中基因表达的变化,这种变化因不同的细胞类型、不同的mRNA含量和不同的细胞状态而表现出多模态和异质性[75]。一项近期的综合基准分析[76]表明,基于负二项混合模型的单细胞差异基因表达方法(如glmmTMB [77]和NEBULA-HL [78])优于其他多种方法。接下来,可以进行功能分析,以推断基因、基因模块/网络或通路在细胞类型水平的功能。通常,这一步包括基因集富集分析和功能富集分析[37]。
scRNA-seq数据库
随着针对多种植物物种的scRNA-seq数据快速积累,迫切需要建立在线数据库,以促进scRNA-seq结果的公共共享和利用。近年来,越来越多专用于植物的scRNA-seq数据库被创建,其中一些列于表2。例如,最近开发了一个名为PlantscRNAdb的数据库,用于分析植物scRNA-seq数据,其中包含来自拟南芥、水稻、番茄和玉米的128种不同细胞类型的26,326个标记基因[79]。另一个名为Plant Single Cell Transcriptome Hub(PsctH)的数据库也被创建,以为植物组织中各种细胞类型提供全面而精确的细胞标记资源和网络工具,其中包括来自拟南芥、花生、水稻、番茄和玉米的9种植物组织/亚组织中的51种细胞类型的98个细胞标记[80]。此外,另一个名为Plant Cell Marker DataBase(PCMDB)的数据库包含6种植物物种(拟南芥、大豆、烟草、水稻、番茄和玉米)中22种组织的263种细胞类型的81,117个细胞标记基因,通过手动整理实验验证的标记基因以及基于整体RNA-seq和scRNA-seq数据的差异表达标记基因,为scRNA-seq数据分析中的细胞类型划分提供支持[81]。
目前,大多数植物scRNA-seq数据库仅包含有限物种(少于7种)的细胞基因表达数据。为了克服这一限制,He等人[82]创建了一个综合性数据库scPlantDB,其中涵盖了约250万个细胞,涉及17种植物物种,有助于多种scRNA-seq数据集的分析和利用。
| Database name | Website | Species | Reference |
|---|---|---|---|
| PlantscRNAdb | PlantscRNAdb | Arabidopsis thaliana, Oryza sativa, Solanum lycopersicum, and Zea mays | [79] |
| 3D Flower Meristem | http://threed-flower-meristem.herokuapp.com/ | A. thaliana | [157] |
| The Plant scRNA-Seq Browser (PscB) | https://www.zmbp-resources.uni-tuebingen.de/timmermans/plant-single-cell-browser/ | A. thaliana | [140] |
| Plant Single-Cell Transcriptome Hub (PsctH) | http://jinlab.hzau.edu.cn/PsctH/ | A. thaliana, Z. mays, O. sativa, Arachis hypogaea, and Solanum lycopersicum | [80] |
| Root Cell Atlas | https://phytozome-next.jgi.doe.gov/tools/scrna/ | A. thaliana | [6] |
| stRNAPal | ST of Vascular Bundle Stem | Populus alba × P. glandulosa clone 84K | [85] |
| PCMDB | PlantCellMarker | A. thaliana, Glycine max, Nicotiana tabacum, O. sativa, S. lycopersicum, and Z. mays | [81] |
| scPlantDB | scPlantDB | A. thaliana, Bombax ceiba, Brassica rapa, Catharanthus roseus, Fragaria vesca, G. max, Gossypium bickii, G. hirsutum, Manihot esculenta, Medicago truncatula, N. attenuata, O. sativa, P. alba var. pyramidalis, P. alba × P. glandulosa, S. lycopersicum, Triticum aestivum, and Z. mays | [6,82] |
表2. 部分scRNA-seq数据库列表
scRNA-seq在植物系统生物学中的应用
植物系统生物学是一个跨学科领域,结合计算和实验方法研究复杂的植物生物系统,其重点在于理解系统的各个组成部分如何相互作用并对系统的整体行为产生贡献[83]。通过对植物细胞转录组的分析、追踪其在不同植物细胞发育阶段的动态变化,以及揭示它们对环境胁迫的响应,可以在细胞水平上揭示发育和胁迫响应机制[21]。在本节中,我们重点介绍了scRNA-seq在植物系统生物学研究中的一些最新应用,如图3所示。
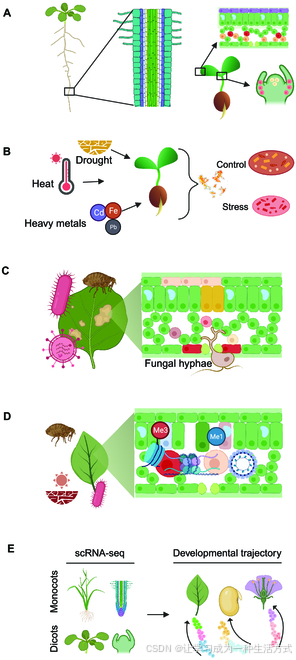
图3. 单细胞RNA测序(scRNA-seq)在植物系统生物学中的一些最新应用 (A) 从根、叶片和茎尖分生组织的细胞类型发育梯度。重新绘制自[7,146]。 (B) 非生物胁迫对细胞水平转录组学的影响。重新绘制自[152]。 (C) 不同生物胁迫对复杂器官中细胞类型的影响。重新绘制自[8]。 (D) 不同环境刺激下细胞类型的表观遗传调控。重新绘制自[153]。Me1:单甲基化;Me3:三甲基化。 (E) 根和茎尖分生组织向不同组织的细胞命运决定。重新绘制自[154]。使用BioRender.com创建。
理解细胞水平上的植物发育分子机制
动态的细胞身份对植物的发育过程至关重要[30]。为了理解植物发育,需要揭示细胞类型、发育领域和调控其分化的调控网络的全貌[84]。scRNA-seq是一种研究细胞特异性转录组学的强大工具。目前,scRNA-seq实验已用于揭示多种植物组织中细胞类型的详细图谱。例如,利用scRNA-seq在黑杨(P. trichocarpa)中鉴定出了茎分化木质部的大部分主要细胞类型[16]。此外,一项结合高分辨率解剖分析和空间转录组学的最新研究,描述了白杨×青杨克隆84K茎中从初生到次生维管组织的分生细胞发育轨迹,并揭示了筛管域中的矩形前形成层样细胞和木质部域中的纺锤状形成层细胞[85]。在拟南芥中,利用scRNA-seq鉴定出了代表地上和地下组织的细胞簇,为特定时间点细胞水平上基因表达的空间调控提供了新见解[86]。
此外,scRNA-seq可以用于研究转录因子在植物发育中的作用。例如,最近利用scRNA-seq生成了玉米根的单细胞分辨率发育图谱,显示出一个高度可移动的转录因子SHORT-ROOT (SHR),该因子调控皮层组织的扩展,在玉米中至少穿越了8层细胞进入皮层[87]。最近的研究还利用scRNA-seq表明,响应于油菜素内酯的两个转录因子HAT7和GTL1与皮层延长相关[88]。
理解植物在细胞水平上的非生物和生物胁迫响应
植物会遭受多种非生物和生物胁迫,例如干旱、洪涝、盐碱、冷冻/低温、高温以及不同病原体的侵害[89]。特定细胞类型对环境条件的响应不同,其中一些细胞类型能够在更严酷的胁迫条件下存活,但目前仍不清楚特定细胞类型如何响应不同的环境信号以及如何与其他细胞沟通以提高生存能力[11]。
scRNA-seq已被用于研究非生物胁迫如何改变植物中细胞类型特异性基因表达。例如,对拟南芥幼苗在热胁迫下的scRNA-seq分析显示,尽管热响应表达在各个细胞类型中主要由经典的热冲击基因主导,但不同细胞类型间其他基因的表达存在细微但显著的差异[13]。此外,对水稻幼苗在高盐、低氮和缺铁等非生物胁迫下地上和地下部分的scRNA-seq分析表明,非生物胁迫主要以细胞类型特异性的方式影响基因表达,而对于特定细胞类型来说,不同胁迫主要调控大致相同的一组基因[15]。
scRNA-seq也被用于研究植物对由细菌、真菌病原体以及昆虫引起的生物胁迫的响应。例如,一项研究利用scRNA-seq分析了受丁香假单胞菌(Pseudomonas syringae)感染的拟南芥叶组织的转录组,揭示了免疫状态、过渡状态和易感状态中表现出转录响应的不同病原体响应细胞簇,为疾病进展的分子机制提供了见解[90]。此外,对拟南芥叶组织的scRNA-seq分析揭示了细胞类型特异性的基因表达对真菌病原体丁香葡萄孢(Colletotrichum higginsianum)的响应,并展示了表皮细胞中表达的MYB122基因在抗病性中的作用[91]。进一步地,比较不同水稻品种对褐飞虱(Nilaparvata lugens)响应的scRNA-seq分析显示,在易感(TN1)和抗性(YHY15)水稻品种中,细胞类型(如中鞘细胞、保卫细胞、叶肉细胞、木质部细胞、泡状细胞和韧皮部细胞)在抗褐飞虱能力方面存在显著差异[92]。
理解植物细胞水平上的表观遗传调控
表观遗传修饰(如DNA甲基化和组蛋白修饰)在调控植物基因表达中起着重要作用。尽管全球基因表达的表观遗传调控已被广泛研究[93,94],但单细胞水平上的表观遗传调控在植物中尚未被深入探索。单细胞表观基因组学及其新兴应用主要在动物中进行研究[95–97]。例如,结合单细胞RNA测序(scRNA-seq)与转座酶可接近染色质测序(ATAC-seq),已用于分析小鼠心脏祖细胞,深入了解心脏祖细胞命运决定过程中的转录和表观遗传调控[98]。类似地,对人类免疫表型血细胞的整合性scRNA-seq和ATAC-seq分析表明,在造血干细胞/多能祖细胞分化为特定谱系之前,发生了广泛的表观遗传而非转录性预编程[99]。受哺乳动物研究的启发,这些策略也可应用于植物单细胞表观基因组学研究。
scRNA-seq可以与表观遗传分析技术(如染色质免疫共沉淀测序[ChIP-seq]、ATAC-seq或亚硫酸盐测序)结合,用于分析基因表达与表观遗传修饰之间的关系[100]。例如,对小麦根样本的单细胞核RNA测序(snRNA-seq)和ATAC-seq的整合分析揭示了不对称基因转录和驱动根毛分化的细胞类型特异性基因调控网络[101]。总体而言,scRNA-seq正逐渐成为揭示植物中基因表达与表观遗传调控关系的强大工具。
理解植物中的细胞命运决定与器官发生
植物细胞命运决定是一个复杂的过程,涉及各种信号通路和调控机制。在生长过程中,细胞分化为特定细胞类型是由特定基因的表达决定的,而这些基因的表达受到各种转录因子和信号分子的控制[102]。scRNA-seq在揭示植物细胞命运决定的分子机制方面具有巨大潜力。例如,最近一项研究使用scRNA-seq对陆地棉(Gossypium hirsutum)胚珠外珠被在四个发育阶段的基因表达进行了分析,鉴定出两个转录因子(MYB25-like和HOX3),分别作为纤维分化和顶端偏向扩展生长的关键调控因子[103]。
此外,对再生型棉花基因型Jin668和非再生型TM-1的下胚轴组织进行的scRNA-seq分析表明,初生维管细胞类型在响应外部刺激时经历细胞命运转变,并鉴定出一些新型再生相关基因,如CSEF、PIS1、AFB2、ATHB2、PLC2和PLT3[104]。为研究植物主要促进横向生长的组织——木质部中的细胞发育,Tung等人[105]对四种植物物种(两种核心真双子叶植物黑杨和大桉,一种基部真双子叶植物厚皮香,以及一种基部被子植物鹅掌楸)进行了比较空间转录组学分析,结合scRNA-seq与激光捕获显微切割转录组学,为不同植物物种中木质部细胞谱系的形成提供了深入理解。
单细胞基因组学在植物合成生物学中的应用
植物合成生物学是一个快速发展的领域,致力于设计和创造新的植物性状[17,106]。这一领域具有颠覆农业、改善生态系统可持续性以及提升植物整体健康的潜力,是一个令人兴奋的研究方向,对社会带来多方面的益处[107]。通过理解植物生物系统复杂的细胞动态,可以在细胞水平上实现精确的生物设计。
在本节中,我们重点介绍了scRNA-seq在植物合成生物学中的应用,包括发现新的细胞特异性分子成分以进行细胞特异性植物生物设计、揭示代谢工程的生物合成途径以及构建植物与微生物之间的新型共生关系,如图4所示。
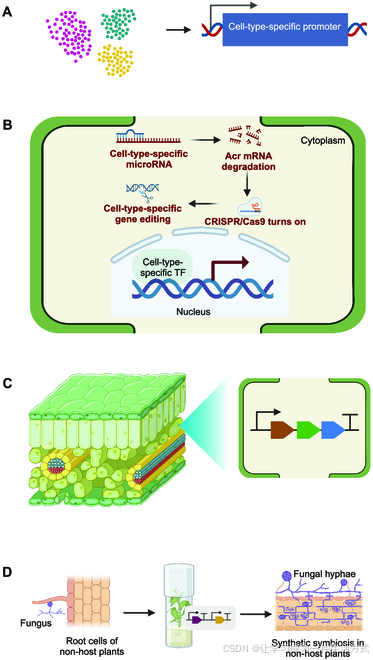
发现和设计用于植物生物工程的细胞类型特异性启动子
从工程的角度来看,细胞类型特异性启动子被定义为能够在特定细胞类型中稳定驱动高水平转基因表达的启动子[108]。植物是包含多种特化细胞类型的多细胞生物,每种细胞类型在器官中具有特定功能和位置[109]。在植物生物工程中,细胞类型特异性启动子相比全时启动子(如CaMV35S)具有显著优势,后者可能因多效性效应对植物产生非预期影响[110,111]。为在植物生物设计中最小化多效性效应的不利影响,需要细胞类型特异性启动子在细胞类型水平上实现基因表达的精确空间控制。
细胞类型特异性启动子广泛应用于植物基因工程[112,113]。具体来说,可以基于通过scRNA-seq分析揭示的植物中具有细胞类型特异性基因表达的基因上游DNA序列(如cis-元件)设计启动子。这些启动子可用于实现细胞类型特异性的基因调控(如过表达或下调)或基因编辑,以工程化特定性状(如细胞类型特异性代谢和发育),同时避免或最小化对非目标性状的影响。例如,利用细胞类型特异性启动子驱动CRISPR/Cas9系统表达,已成功实现细胞类型特异性基因编辑[114,115]。为减少由木质素生物合成抑制基因的过表达引起的生长缺陷,使用木质部纤维特异性启动子在纤维细胞中表达抑制基因[116]。因此,发现和设计更多细胞类型特异性启动子将有助于植物生物设计的进步。
细胞类型特异性启动子的鉴定通常需要在细胞水平上的基因表达分析[117]。scRNA-seq可以识别细胞类型特异性基因表达,为克隆或重新设计细胞类型特异性启动子提供基因组信息[14]。例如,在黑杨茎分化木质部的scRNA-seq分析中发现了两个导管和纤维特异性基因(PtWAT1和PtCesA8),通过在转基因毛白杨中以这些启动子驱动β-葡糖醛酸酶(GUS)报告基因表达进行验证[16]。最近,通过scRNA-seq分析在多种植物物种中发现了许多新的细胞类型和细胞标记基因[118,119],促进了新型细胞类型特异性启动子的设计。此外,与染色质分析方法(如ATAC-seq)结合,scRNA-seq能够发现基因组中的顺式调控元件(CREs)[14,120]。例如,基于scRNA-seq数据,在拟南芥根细胞和玉米不同器官中发现了大量CREs[14,121]。这些细胞类型特异性CREs为构建合成细胞类型特异性启动子提供了宝贵的组件。
设计植物天然产物工程策略
植物系统正成为高产复杂植物天然产物(PNPs)的重要底盘平台[127]。在异源生产PNPs中,鉴定生物合成途径中的关键酶至关重要[127]。scRNA-seq通过分析细胞水平的转录组学,能够探索植物中特化代谢的多细胞分区[128]。例如,对长春花(Catharanthus roseus)中单萜吲哚生物碱(MIA)生物合成途径的scRNA-seq分析显示,MIA的生物合成始于内部筛管相关薄壁细胞,随后途径的酶促步骤优先发生在表皮细胞中,最后步骤发生在独立细胞中[128]。此外,还检测到不同细胞类型间的中间体转运蛋白[128]。
另一个近期的应用案例是通过scRNA-seq在红豆杉(Taxus mairei)茎中创建了单细胞转录组图谱,并结合质谱成像技术,鉴定了控制与紫杉醇生物合成相关的细胞特异性基因表达的多个基因,为紫杉醇生产工程化提供了新知识[129]。因此,scRNA-seq在阐明PNPs的生物合成、转运和储存的空间分布方面具有巨大潜力,为细化植物中的代谢途径以提高PNPs产量提供了详细指导。
设计和工程化固氮根瘤共生
固氮土壤细菌(称为根瘤菌)能够通过共生关系为豆科植物提供稳定的氮源[130]。这种共生关系,也称为结瘤,是从细菌感染根毛细胞开始的,这会诱导根皮层、内皮层和中柱细胞形成根瘤原基,从而形成一种新的根器官——根瘤[130]。自从发现与固氮根瘤菌的这种内共生关系以来,人们对将这一性状转移到非豆科作物中的兴趣浓厚[131]。工程化固氮根瘤共生最可行的方法可能是通过基因转移将结瘤物种的共生基因传递到非结瘤物种中,以模拟已建立的共生关系[131]。植物合成生物学在通过协调遗传编程以实现多个方面(如Nod因子识别、细菌感染、根瘤形成以及在根瘤内创造适合氮酶活性的环境)的固氮根瘤共生工程化方面具有巨大潜力[132]。
受到多种固氮共生形式的启发,固氮根瘤共生的工程化涵盖了多种生化途径和细胞类型[131]。迄今为止,通过组学和功能基因组学研究已经鉴定出了一些共生核心基因[133,134]。然而,由于整体组织水平转录组学分析的分辨率有限,固氮根瘤共生发展的精确图景仍不清楚。scRNA-seq在捕获控制结瘤过程的细胞水平转录组程序方面具有巨大潜力。最近,对紫花苜蓿(Medicago truncatula)根系接种后早期阶段的scRNA-seq分析建立了一个高分辨率空间转录组图谱,发现了许多控制紫花苜蓿根对根瘤菌接种响应的新基因和功能通路[130]。
scRNA-seq在植物中的应用面临的挑战与潜在解决方案
尽管scRNA-seq在植物系统和合成生物学研究中具有巨大前景,但在样本制备、文库构建、数据分析以及数据共享和利用方面仍存在技术挑战。植物细胞被坚硬的细胞壁包围,这可能影响完整细胞核或原生质体的分离以进行scRNA-seq。高效的细胞解离方法仍需要进一步开发,以用于制备细胞核或原生质体样本[135]。原生质体的产量和活性受到组织质量、基因型、生理状态以及胁迫响应等多种因素的影响。一个解决方案是优化分离条件,包括酶处理时间、温度和渗透势,以提高原生质体的产量和活性[136]。
相比于scRNA-seq,snRNA-seq在分析复杂植物组织时具有优势,特别是在植物细胞壁对原生质体分离构成挑战时[137]。最近的研究报道了与高通量snRNA-seq兼容的高质量细胞核分离协议[137]。然而,由于植物细胞核体积较小且易受损,分离完整细胞核仍然具有挑战性。维持细胞核在分离过程中的完整性需要仔细处理并优化裂解缓冲液的成分和离心参数[138]。
在文库构建中,广泛使用的基于液滴的方法(如10x Chromium方法)存在一个技术挑战,即在某些情况下,一个液滴中可能包含多个细胞或细胞核,或者包含次优数量的珠子。因此,未来需要进一步的技术改进以确保一个液滴中仅包含一个珠子和一个细胞或细胞核[139]。
植物细胞较大的体积对scRNA-seq研究提出了潜在挑战,目前尚未有理想的细胞制备方法被报道[140]。一种潜在的解决方案是将一些在动物系统中开发的新型scRNA-seq技术应用于植物。例如,最近报道了一种名为RevGel-seq的scRNA-seq方法突破性进展,它在哺乳动物细胞样本制备过程中无需特定的单细胞RNA设备[141]。传统的scRNA-seq样本制备依赖于专用且仪器依赖的方法,而RevGel-seq基于细胞条形码珠子复合物,在人类和小鼠细胞中无需特定仪器即可实现便捷高效的样本制备。研究人员通过与10x Chromium方法使用相同的数据分析平台进行比较,鉴定出了相同的细胞类型并具有一致的丰度等级。这表明RevGel-seq可能成为10x Chromium方法的可行替代方案,尽管尚未在植物细胞中进行测试。RevGel-seq还具有允许在样本制备过程中提前终止的优势,从而在样本收集和不同时间或地点的处理方面提供灵活性。
其他在动物系统中开发的、更廉价的技术也可能应用于植物,例如分体式池连接转录组测序(SPLiT-seq),该方法通过组合条形码分析单细胞转录组,而无需物理分离每个细胞[142,143];以及基于颗粒模板的即时分区测序技术,无需微流体设备[144]。此外,一种名为“通过dA尾加长进行单细胞广泛转录组分析”(VASA-seq)的新方法最近被开发,用于在小鼠中捕获非聚腺苷化和聚腺苷化转录本全长的数据[145]。与转录本3′端测序相比,VASA-seq在植物单细胞样本中的应用潜力巨大,可提供更全面的转录信息。
非模式物种中缺乏充分表征的细胞类型特异性标记基因是scRNA-seq数据分析的主要挑战[143]。目前已取得新进展来解决这一问题,例如开发了包含81,117个标记基因的PCMDB数据库,该数据库覆盖了6种植物的22种组织的263种细胞类型[81]。
另一个挑战是许多植物物种的参考基因组不完整或注释不充分,这可能阻碍scRNA-seq数据的精确比对和解释。细胞间异质性需要生成大规模数据集并利用强大的生物信息学流程,以准确识别不同的细胞类型[146]。然而,目前缺乏基于来自多种植物物种的scRNA-seq数据进行细胞类型同源性和多样性分类的计算方法[136]。一个潜在的解决方案是建立一个标准化的植物空间单细胞基因组学数据库,整合最新的T2T(从端粒到端粒)基因组、基因注释以及来自多个物种的scRNA-seq数据,涵盖不同发育阶段和环境条件,并采用标准化格式[147]。
结论与展望
单细胞RNA测序(scRNA-seq)已成为一项突破性技术,在植物系统生物学和合成生物学中具有广泛的应用。高质量原生质体或细胞核样本的制备是scRNA-seq成功的关键。植物中scRNA-seq分析选择使用原生质体还是细胞核取决于研究目标、植物物种以及转录组覆盖范围、空间信息和技术可行性之间的平衡。原生质体提供了转录组的全面视角,而细胞核分离方法专注于核编码RNA转录本。然而,由于细胞壁消化困难,从木本植物中分离原生质体和细胞核是非常具有挑战性的。未来的研究应优先开发优化的原生质体分离方法,特别是针对木本植物。
正如图5A所示,scRNA-seq在植物系统生物学研究中的未来应用蕴含着许多机会。随着scRNA-seq在全球范围内植物系统生物学和合成生物学研究中的迅速推广,各实验室和植物物种间的scRNA-seq流程(包括样本制备、文库构建、测序和数据分析)标准化需求迫在眉睫。目前,关于标准scRNA-seq流程的制定工作已经展开[148]。开发标准化协议和质量控制措施将确保scRNA-seq数据的可重复性和可比性,从而实现更强大的分析和更可靠的结论。
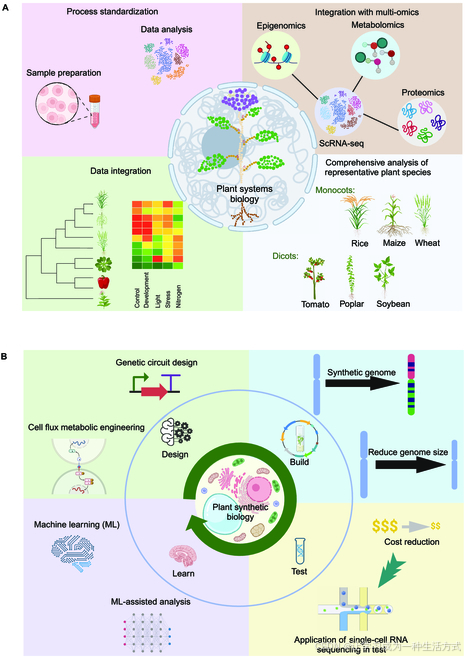
图5. 单细胞RNA测序(scRNA-seq)在植物系统生物学和合成生物学中的应用展望 (A) scRNA-seq在植物系统生物学中的应用机会,包括scRNA-seq流程的标准化、与其他组学技术的整合、在多种植物物种中的应用以及数据整合。 (B) scRNA-seq在植物合成生物学“设计–构建–测试–学习”(DBTL)循环中的应用机会,包括设计遗传电路、构建合成基因组、测试工程系统以及为下一轮DBTL学习新知识。使用BioRender.com创建。
整合scRNA-seq与其他组学技术
将scRNA-seq与其他组学技术整合是一个有前景的发展方向。例如,最近一项报告将scRNA-seq与ChIP-seq/ATAC-seq整合,用于构建玉米(Z. mays)的转录网络[84]。多种计算工具已被开发用于异质单细胞多组学数据的综合分析,例如GLUE(图链接统一嵌入)[149]和Con-AAE(对比循环对抗自编码器)[150]。多组学数据整合策略将提升对植物细胞过程和调控网络的全面理解,使研究人员能够揭示植物生物学在系统水平上复杂的相互作用和机制。
扩展scRNA-seq在非模式植物中的应用
scRNA-seq的应用需要扩展到更多种类的植物物种,而不仅限于研究较多的模式生物。这不仅能够揭示特定植物物种中的生物学过程,还能有助于发现整个植物界中保守的调控网络。需要进一步开发数据整合和分析方法,以处理来自多种植物物种和实验条件的大量scRNA-seq数据。
scRNA-seq在植物合成生物学中的未来角色
未来,scRNA-seq将在推动植物合成生物学发展中发挥重要作用,通过生成高分辨率基因表达数据,增强植物合成生物学的“设计–构建–测试–学习”(DBTL)能力,如图5B所示。这将为细胞水平上的生物学过程和基因表达调控提供前所未有的新见解,可用于提高植物基因工程的效率、可预测性和稳定性。研究人员可以利用scRNA-seq数据设计合成基因组或减少基因组大小,从而为合成生物学应用解锁新的可能性。
利用机器学习辅助分析scRNA-seq数据,可以深入了解单个基因的功能角色及其相互作用,从而设计更精确高效、适用于特定应用的合成基因组。到目前为止,由于这一新技术的高成本,scRNA-seq尚未被广泛用于测试工程化植物。然而,预计未来的技术进步将显著降低scRNA-seq分析的成本,从而使越来越多的研究人员能够在细胞水平上研究转基因植物的基因表达。