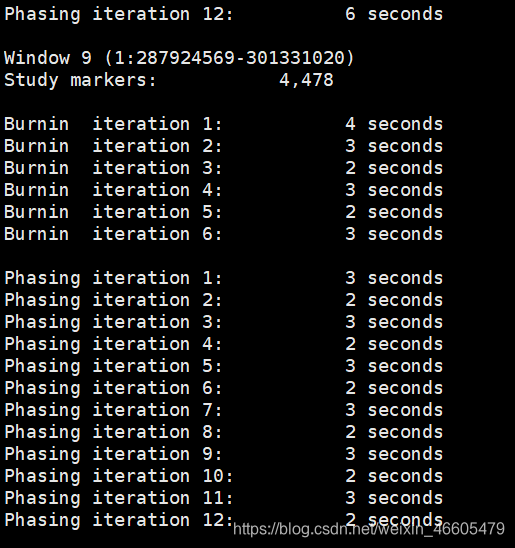
在使用BEAGLE5.0版本填补基因型时,遇到这样一个问题:
接着在网上搜索了一下解决办法,在biostar上看到有人遇到了同样的问题,有人建议使用BEAGLE4.0版本,我尝试后仍然没有解决问题。然后多方搜索发现有这样一个帖子:
网址如下:https://ask.csdn.net/questions/4419232
这个问题应该和我的类似,我的vcf文件里面多了“-”符号,而这个帖子里多了“*”。
于是数了一下文件里有多少行“-”:
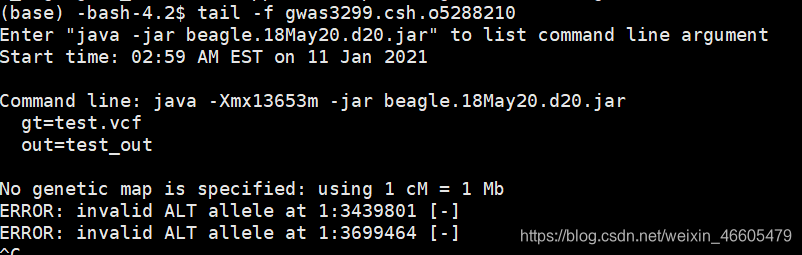
find test.vcf | xargs cat | grep .*-.*|wc –l
接着把含有“-”符号的行删掉:
sed -e "/-/d" out.vcf > test_out.vcf
现在已经解决了。