RNA-seq数据的批次校正方法
bulk-RNA seq过程可能存在不同建库批次以及不同测序深度带来的如测序深度等batch effects,那么该如何整合这些数据。
Batch effects
批次效应是在处理样本过程中因为技术因素引入的变量。通常我们不可能一次性由同一个人或同一台测序仪对所有样本进行测序,不同批次的样本它们之间可能存在较大的技术差异,这些差异构成了批次效应。技术因素导致的差异可能会对我们后期分析生物学差异产生较大的影响,因此如何将降低这些非研究的因素引发的批次效应是很有意义的。
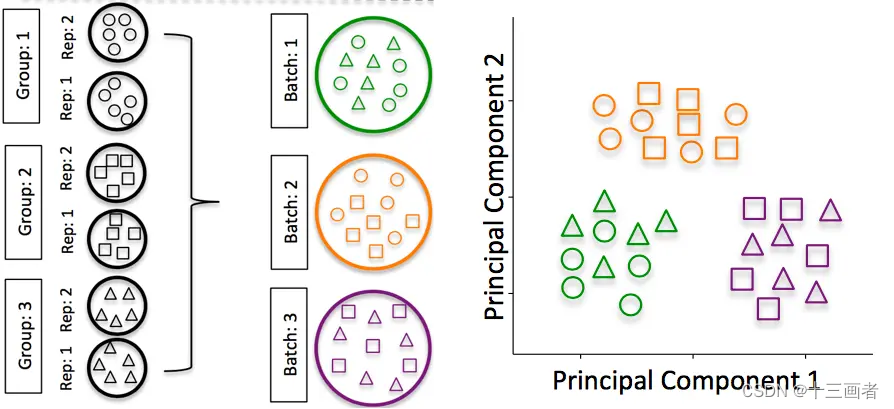
一般在实验设计之初,研究人员就应该考虑到如何避免引入可能混淆生物学意义的Technical batch effects(比如在肠道微生物研究领域,因为不同年龄段的肠道微生物存在较明显的差异,如果做case/control的研究,我们会控制两组人群的年龄分布无显著差异,除此之外如性别等也会考虑)。一项研究是否合理,前期实验设计尤其重要。
批次校正只能降低批次效应的影响,而不能完全消除批次效应,因此使用 reduce the batch effect 而不是delete the batch effect。在假定实验设计没有问题时,可以通过探究数据结构的方法去评估批次效应,衡量的方法如: 1. distance measures(距离法);2. clustering(样品层次聚类法); 3.spatial methods(空间统计法)。在识别数据存在批次效应后,可以通过设置biological variables 和 corrected variables 进行线性回归校正批次效应。
Batch effects are sub-groups of measurements that have qualitatively different behaviour across conditions and are unrelated to the biological or scientific variables in a study. (Leek et,al 2010)
校正步骤
根据批次设置不同颜色的分组,然后根据一下步骤进行:
-
使用PCA或层次聚类对所有样本进行可视化;
-
对生物学意义分组进行区别,查看biological treatment的样本分布;
-
再根据不同来源的Technical batch effects对生物学意义的影响,例如采样时间、建库时间和测序平台等等;
-
处理校正批次效应的三种方式:
1. 不做任何处理,但在后续分析应该意识到批次效应的存在可能对组内差异结果有某种程度的贡献,当然也可能导致无法找到组间差异; 2. 试图降低批次效应,这意味着需要对数据进行处理和转换,该过程即可能会移除技术差异也可能移除组间差异,这是一个需要考虑的过程,当然在降低批次效应后,组间比较的结果可能更具生物学意义和统计效能; 3. 根据批次去做同一批次分析,最后再结合meta analysis找到某些variable是否在不同批次均出现,该方法因为每个batch的sample size太小会导致统计power降低,好处就是不需要转换数据。
现在针对来源不同的bulk RNA seq数据有了很多不同的方法或R包校正批次效应,这里我通过实例介绍几类常用的R包
实例
数据预处理
从EBI下载了3个不同批次的小鼠 bulk-RNA raw data (fastq),经过RNA-seq pipeline处理后,我们获得了这些数据集的基因表达counts数据。因为数据只有不同批次和疾病分组两类表型信息,所以只能设置批次为校正变量和疾病为生物学处理变量。
在处理前,先对数据进行过滤处理以及存成ExpressionSet格式的数据对象。
ExpressionSet 是 Biobase 包提供的。Biobase 本身是 Bioconductor 项目的一个部分,为很多基因组学数据提供数据类型支持。ExpressionSet 就是用来专门将多种不同来源的数据整合到一起方便数据处理的一种数据类型,很多 Bioconductor 函数输入输出的都是 ExpressionSet 类型的数据。
简单的说,ExpressionSet 把表达数据(
assayData存储芯片、测序等表达数据),表型信息(phenoData存储样本信息),注释等元信息(featureData,annotation存储芯片或者测序技术的元数据),以及操作流程(protocolData存储样本处理相关的信息,通常由厂家提供)和实验(experimentData用来描述实验相关信息)几种密切相关数据封装在一起,这样我们处理数据的时候不用关心各个数据的细节把它们当作一个整体来看就好。
step1: loading data
library(dplyr)
library(data.table)
library(tibble)
library(convert)
library(ggplot2)
rm(list = ls())
options(stringsAsFactors = F)
options(future.globals.maxSize = 1000 * 1024^2)
mus_GSE1<- fread("mus_GSE1_stringtie_counts.csv")
mus_GSE2 <- fread("mus_GSE2_stringtie_counts.csv")
mus_GSE3 <- fread("mus_GSE3_stringtie_counts.csv")
# phenotype : sampleid, batch, disease
phen <- read.csv("public_data_phenotype_20201120.csv")
step2: Create ExpressionSet
get_expr_Set <- function(x=phen, y=mus.prf.lst, ncount=10, occurrence=0.2){
# x=phen
# y=mus.prf.lst
# ncount=10
# occurrence=0.2
prof <- y[[1]]
for(i in 2:length(y)){
prof <- inner_join(prof, y[[i]], by="gene_id")
}
prof$gene_id <- gsub("\\|\\S+", "", prof$gene_id)
prf <- prof %>% column_to_rownames("gene_id")
sid <- intersect(x$SampleID, colnames(prf))
phe <- x %>% filter(SampleID%in%sid) %>%
mutate(Batch=factor(as.character(Batch)),
Group=factor(as.character(Group))) %>%
mutate(batch_number=as.numeric(Batch)) %>%
column_to_rownames("SampleID")
prf.cln <- prf %>% rownames_to_column("tmp") %>%
filter(apply(dplyr::select(., -one_of("tmp")), 1, function(x) {
sum(x != 0)/length(x)}) > occurrence) %>%
dplyr::select(c(tmp, rownames(phe))) %>%
column_to_rownames("tmp")
temp <- apply(prf.cln, 1, function(x){length(unique(x[x>0]))}) %>%
data.frame() %>% setNames("number")
remain_genes <- temp %>% filter(number > 2)
prf.cln <- prf.cln[rowSums(prf.cln) > ncount, ]
prf.cln2 <- prf.cln[rownames(prf.cln)%in%rownames(remain_genes), ]
# determine the right order between profile and phenotype
for(i in 1:ncol(prf.cln2)){
if (!(colnames(prf.cln2)[i] == rownames(phe)[i])) {
stop(paste0(i, " Wrong"))
}
}
exprs <- as.matrix(prf.cln2)
adf <- new("AnnotatedDataFrame", data=phe)
experimentData <- new("MIAME",
name="Hua Zou", lab="Bioland Lab",
contact="[email protected]",
title="Experiment",
abstract="The gene ExpressionSet",
url="www.grmh-gdl.cn",
other=list(notes="Created from text files"))
expressionSet <- new("ExpressionSet", exprs=exprs,
phenoData=adf,
experimentData=experimentData)
return(expressionSet)
}
pca_fun <- function(expers_set=mus.set){
# expers_set=mus.set
pheno <- pData(expers_set)
edata <- exprs(expers_set)
# normalizing data by scale
pca <- prcomp(t(edata), scale. = TRUE, center = T)
score <- inner_join(pca$x %>% data.frame() %>%
rownames_to_column("SampleID") %>%
select(c(1:3)) ,
pheno %>% rownames_to_column("SampleID"),
by = "SampleID")
pl <- ggplot(score, aes(x=PC1, y=PC2))+
geom_point(aes(color=Batch),
size=3.5)+
theme_bw()
return(pl)
}
step3: Detect the batch effects
mus.prf.lst <- list(mus_GSE1, mus_GSE2, mus_3)
mus.set <- get_expr_Set(y=mus.prf.lst)
saveRDS(mus.set, "./ReduceBatchEffect/mus.origin.RDS", compress = TRUE)
# hierarchical clustering
pheno <- pData(mus.set)
edata <- exprs(mus.set)
dist_mat <- dist(t(edata))
clustering <- hclust(dist_mat, method = "complete")
par(mfrow=c(2,1))
plot(clustering, labels = pheno$Batch)
plot(clustering, labels = pheno$Group)
# PCA: 在计算协方差时候需要标准化数据,通常使用scale中心化数据
pca_fun(expers_set = mus.set)
SVA + ComBat_seq
SVA包的开发版本增加了最新的ComBat_seq函数,相比之前的ComBat函数,ComBat_seq是基于ComBat函数基础针对RNA-seq count数据开发的工具,它使用了**negative binormial regression(负二项回归)**处理count矩阵。两者都可以处理已知 Batch effects和潜在的batch effects。上bioconductor安装最新的SVA包.
校正模型的方法是构建线性模型。基础使用代码:
library(sva)
count_matrix <- matrix(rnbinom(400, size=10, prob=0.1), nrow=50, ncol=8)
batch <- c(rep(1, 4), rep(2, 4))
# one factor
adjusted <- ComBat_seq(count_matrix, batch=batch, group=NULL)
# one biological variable
group <- rep(c(0,1), 4)
adjusted_counts <- ComBat_seq(count_matrix, batch=batch, group=group)
# multiple biological variables
cov1 <- rep(c(0,1), 4)
cov2 <- c(0,0,1,1,0,0,1,1)
covar_mat <- cbind(cov1, cov2)
adjusted_counts <- ComBat_seq(count_matrix, batch=batch, group=NULL, covar_mod=covar_mat)
step1:在设置multiple biological variable过程报错,后续可能需要再次调整
library(sva)
ComBat_seq_fun <- function(x=mus.set){
# x=mus.set
qcMetadata <- pData(x)
qcData <- t(exprs(x)) %>% data.frame()
# determine the right order between profile and phenotype
for(i in 1:nrow(qcData)){
if (!(rownames(qcData)[i] == rownames(qcMetadata)[i])) {
stop(paste0(i, " Wrong"))
}
}
# covar_mat <- model.matrix(~Group,
# data=qcMetadata)[, -1]
# colnames(covar_mat) <- gsub("Group", "", colnames(covar_mat))
# colnames(covar_mat) <- gsub("Batch", "", colnames(covar_mat))
# adjusted_counts <- ComBat_seq(t(qcData),
# batch=qcMetadata$Batch,
# group=NULL,
# covar_mod=covar_mat)
adjusted_counts <- ComBat_seq(t(qcData),
batch=qcMetadata$Batch,
group=NULL)
exprs <- as.matrix(adjusted_counts)
adf <- new("AnnotatedDataFrame", data=qcMetadata)
experimentData <- new("MIAME",
name="Hua Zou", lab="Bioland Lab",
contact="[email protected]",
title="Experiment",
abstract="The gene ExpressionSet",
url="www.grmh-gdl.cn",
other=list(notes="ajusted counts by ComBat_seq"))
expressionSet <- new("ExpressionSet", exprs=exprs,
phenoData=adf,
experimentData=experimentData)
return(expressionSet)
}
step2: 查看校正前后的结果
mus.set.combatseq <- ComBat_seq_fun(x = mus.set)
cowplot::plot_grid(pca_fun(expers_set = mus.set),
pca_fun(expers_set = mus.set.combatseq),
ncol = 2,
labels = c("origin", "combatseq"))
saveRDS(mus.set.combatseq, "./ReduceBatchEffect/mus.combatseq.RDS",
compress = TRUE)
Result : 校正结果不理想,可能是没有设置好cov.mat的原因,或许我应该尝试ComBat。
limma+removeBatchEffect
该函数最开始针对芯片数据设计,我在应用该函数时候没有考虑到该因素,导致输入的是count data,最后返回的结果没有任何的变化,因此是错误的示范。输入数据应该是标准化后的数据(如 log化),或者是DESeq2量化因子后的数据。此处是错误的示范。
step1: 构建函数
library(limma)
removeBatchEffect_fun <- function(x=mus.set){
qcMetadata <- pData(x)
qcData <- exprs(x)
design <- model.matrix(~Group+Batch,
data=qcMetadata)
colnames(design) <- gsub("Batch", "", colnames(design))
colnames(design) <- gsub("Group", "", colnames(design))
limma.edata <- removeBatchEffect(qcData,
batch = qcMetadata$Batch,
design = design)
exprs <- as.matrix(limma.edata)
adf <- new("AnnotatedDataFrame", data=qcMetadata)
experimentData <- new("MIAME",
name="Hua Zou", lab="Bioland Lab",
contact="[email protected]",
title="Experiment",
abstract="The gene ExpressionSet",
url="www.grmh-gdl.cn",
other=list(notes="ajusted counts by removeBatchEffect"))
expressionSet <- new("ExpressionSet", exprs=exprs,
phenoData=adf,
experimentData=experimentData)
return(expressionSet)
}
step2: 结果
mus.set.rbe <- removeBatchEffect_fun(x = mus.set)
cowplot::plot_grid(pca_fun(expers_set = mus.set),
pca_fun(expers_set = mus.set.rbe),
ncol = 2,
labels = c("origin", "removeBatchEffect"))
saveRDS(mus.set.rbe, "./ReduceBatchEffect/mus.removeBatchEffect.RDS",
compress = TRUE)
VOOM+SNM
VOOM根据分组进行标准化数据,SNM是一类有监督的标准化方法,前者可以降低测序深度的影响,后者则可以降低批次效应的影响,两个结合使用更利于校正批次效应。
step1: 构建函数
library(limma)
library(edgeR)
library(snm)
library(gbm)
VoomSNM_fun <- function(x=mus.set){
qcMetadata <- pData(x)
qcData <- t(exprs(x))
# Set up design matrix
covDesignNorm <- model.matrix(~0 + Batch + Group,
data = qcMetadata)
# The following corrects for column names that are incompatible with downstream processing
colnames(covDesignNorm) <- gsub('([[:punct:]])|\\s+','',colnames(covDesignNorm))
# Set up counts matrix
counts <- t(qcData) # DGEList object from a table of counts (rows=features, columns=samples)
# Quantile normalize and plug into voom
dge <- DGEList(counts = counts)
vdge <- voom(dge, design = covDesignNorm, plot = TRUE, save.plot = TRUE,
normalize.method="none")
# List biological and normalization variables in model matrices
bio.var <- model.matrix(~Group,
data=qcMetadata)
colnames(bio.var) <- gsub('([[:punct:]])|\\s+','',colnames(bio.var))
adj.var <- model.matrix(~Batch,
data=qcMetadata)
colnames(adj.var) <- gsub('([[:punct:]])|\\s+','',colnames(adj.var))
snmDataObjOnly <- snm(raw.dat = vdge$E,
bio.var = bio.var,
adj.var = adj.var,
rm.adj=TRUE,
verbose = TRUE,
diagnose = TRUE)
snmData <- snmDataObjOnly$norm.dat
exprs <- as.matrix(snmData)
adf <- new("AnnotatedDataFrame", data=qcMetadata)
experimentData <- new("MIAME",
name="Hua Zou", lab="Bioland Lab",
contact="[email protected]",
title="Experiment",
abstract="The gene ExpressionSet",
url="www.grmh-gdl.cn",
other=list(notes="ajusted counts by removeBatchEffect"))
expressionSet <- new("ExpressionSet", exprs=exprs,
phenoData=adf,
experimentData=experimentData)
return(expressionSet)
}
step2: 结果
mus.set.VoomSNM <- reduce_voom_SNM(x = mus.set)
cowplot::plot_grid(pca_fun(expers_set = mus.set),
pca_fun(expers_set = mus.set.VoomSNM),
ncol = 2,
labels = c("origin", "VoomSNM"))
saveRDS(mus.set.VoomSNM, "./ReduceBatchEffect/mus.VoomSNM.RDS",
compress = TRUE)
DESeq2
DESeq2包采用
DESeqDataSet存储原始的read count和中间计算的统计量。每个
DESeqDataSet对象都要有一个实验设计formula,用于对数据进行分组,以便计算表达值的离散度和估计表达倍数差异,通常格式为~ batch + conditions(为了方便后续计算,最为关注的分组信息放在最后一位)。
countData: 表达矩阵
colData: 样品分组信息表
design: 实验设计信息,conditions必须是colData中的一列
DESeq2提出的量化因子标准化方法已经考虑到不同批次的样本可能存在批次效应的问题。
**量化因子 (size factor, SF)**是由
DESeq提出的。其方法是首先计算每个基因在所有样品中表达的几何平均值。每个细胞的量化因子(size factor)是所有基因与其在所有样品中的表达值的几何平均值的比值的中位数。由于几何平均值的使用,只有在所有样品中表达都不为0的基因才能用来计算。这一方法又被称为 RLE (relative log expression)。
# create DESeqDataSet
pheno <- pData(mus.set)
edata <- exprs(mus.set)
ddsFullCountTable <- DESeqDataSetFromMatrix(countData = t(edata),
colData = pheno, design= ~ conditions)
dds <- DESeq(ddsFullCountTable)
# noramlized Data
normalized_counts <- counts(dds, normalized=TRUE)
# ranked by mad value
normalized_counts_mad <- apply(normalized_counts, 1, mad)
normalized_counts <- normalized_counts[order(normalized_counts_mad, decreasing=T), ]
# log matrix
rld <- rlog(dds, blind=FALSE)
rlogMat <- assay(rld)
rlogMat <- rlogMat[order(normalized_counts_mad, decreasing=T), ]
# clustering
pearson_cor <- as.matrix(cor(rlogMat, method="pearson"))
hc <- hcluster(t(rlogMat), method="pearson")
heatmap.2(pearson_cor,
Rowv=as.dendrogram(hc),
symm=T,
trace="none",
col=hmcol,
margins=c(11,11),
main="The pearson correlation of eachsample")
# downstream analysis : remove batch effects
ddsFullCountTable <- DESeqDataSetFromMatrix(countData = edata,
colData = pheno, design= ~ batch + conditions)
dds <- DESeq(ddsFullCountTable)
rld <- rlog(dds, blind=FALSE)
rlogMat <- assay(rld)
rlogMat <- limma::removeBatchEffect(rlogMat, c(pheno$batch))
# sva reduce the potential batch effects
dat <- counts(dds, normalized=TRUE)
idx <- rowMeans(dat) > 1
dat <- dat[idx, ]
mod <- model.matrix(~ dex, colData(dds))
mod0 <- model.matrix(~ 1, colData(dds))
# calculating the variables
n.sv <- num.sv(dat, mod, method="leek") #gives 11
# using 4
lnj.corr <- svaBatchCor(dat, mod, mod0, n.sv=4)
co <- lnj.corr$corrected
总结
每一种校正方法均有其特色,根据自己数据的特点选择适合数据的校正方法。
系统信息
sessionInfo()
R version 4.0.2 (2020-06-22)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19042)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.1252 LC_CTYPE=English_United States.1252
[3] LC_MONETARY=English_United States.1252 LC_NUMERIC=C
[5] LC_TIME=English_United States.1252
system code page: 936
attached base packages:
[1] parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] data.table_1.13.2 gbm_2.1.8 snm_1.36.0 edgeR_3.30.3
[5] ggplot2_3.3.2 sva_3.35.2 BiocParallel_1.22.0 genefilter_1.70.0
[9] mgcv_1.8-33 nlme_3.1-150 convert_1.64.0 marray_1.66.0
[13] limma_3.44.3 Biobase_2.48.0 BiocGenerics_0.34.0 tibble_3.0.3
[17] dplyr_1.0.2
loaded via a namespace (and not attached):
[1] bit64_0.9-7 jsonlite_1.7.1 splines_4.0.2 statmod_1.4.34
[5] stats4_4.0.2 blob_1.2.1 yaml_2.2.1 pillar_1.4.6
[9] RSQLite_2.2.0 lattice_0.20-41 glue_1.4.2 digest_0.6.25
[13] minqa_1.2.4 colorspace_1.4-1 cowplot_1.1.0 htmltools_0.5.0
[17] Matrix_1.2-18 XML_3.99-0.5 pkgconfig_2.0.3 purrr_0.3.4
[21] xtable_1.8-4 corpcor_1.6.9 scales_1.1.1 lme4_1.1-25
[25] annotate_1.66.0 generics_0.1.0 farver_2.0.3 IRanges_2.22.2
[29] ellipsis_0.3.1 withr_2.3.0 survival_3.2-7 magrittr_1.5
[33] crayon_1.3.4 memoise_1.1.0 evaluate_0.14 MASS_7.3-53
[37] rsconnect_0.8.16 tools_4.0.2 lifecycle_0.2.0 matrixStats_0.57.0
[41] stringr_1.4.0 S4Vectors_0.26.1 munsell_0.5.0 locfit_1.5-9.4
[45] AnnotationDbi_1.50.3 compiler_4.0.2 rlang_0.4.7 grid_4.0.2
[49] RCurl_1.98-1.2 nloptr_1.2.2.2 rstudioapi_0.11 bitops_1.0-6
[53] labeling_0.4.2 rmarkdown_2.5 boot_1.3-25 gtable_0.3.0
[57] DBI_1.1.0 R6_2.5.0 knitr_1.30 bit_4.0.3
[61] stringi_1.5.3 Rcpp_1.0.5 vctrs_0.3.4 tidyselect_1.1.0
[65] xfun_0.18