生信碱移
因果 WGCNA分析
CWGCNA结合了中介模型和 WGCNA,以发现 WGCNA 模块、模块特征和表型之间的因果关系,从而揭示模块变化是表型产生的原因还是结果。
随着下一代测序技术的深入应用,高通量组学数据的分析方法层出不穷。其中,WGCNA(加权基因共表达网络分析)是最受欢迎的工具之一。正式分析中,WGCNA 首先通过软阈值策略将基因分组为高共表达的模块,这既避免了信息丢失,又增强了结果的稳健性。随后,WGCNA 通过模块与表型性状的相关性,分析与表型相关的特征(比如基因)模块。尽管如此,WGCNA 只能分析模块与表型是否关联,而无法分析这种关联过程中,模块与表型的因果关联(谁是原因?谁是结果?)。
在此基础上,来自美国国家癌症研究所的研究者开发了R包:因果WGCNA (CWGCNA,causal WGCNA),于今年 4 月份发表在 NAR Genomics and Bioinformatics [IF: 4.60]。它基于传统的 WGCNA 流程,但挖掘了更多信息。CWGCNA 在表型、WGCNA模块和模块特征之间进行「中介分析」,使得WGCNA揭示的无向相关性得以明确化为有向关系,即找到它们的因果关联。更明确的说,CWGCNA 旨在证明究竟是共表达模块变化引起表型发生变化,还是表型变化导致了共表达模块发生变化。

▲ DOI: 10.1093/nargab/lqae042
CWGCNA的核心是中介分析,这是一种用于理解机制并识别干预点的方法。它评估一个变量(暴露)对另一个变量(结果)的影响在多大程度上被某个中介变量所介导。具体来说,CWGCNA 利用 WGCNA 模块、模块特征和表型构建中介模型。对于 RNA 表达数据,模块指的是转录谱高度相关的一组基因,特征是该组中的基因。结合limma包的差异分析(回归方法),中介模型利用模块的特征主成分(PC1值,即模块中所有基因的第一主成分)、基因表达水平和表型性状来测试两种因果方向:① “共表达模块→模块特征→表型”(正向),这表示共表达模块通过其特征驱动表型;② “表型→模块特征→模块”(反向),这表示模块只是由于表型的变化而发生变化。通过这些测试,CWGCNA 可以明确表型与模块之间的因果联系及中介机制。
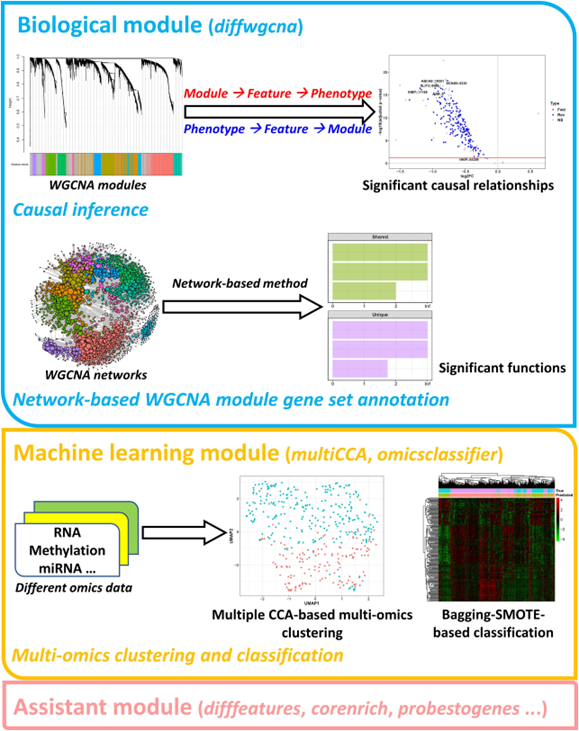
▲ CWGCNA分析的整体流程
不仅如此,CWGCNA 还包含其他用于组学分析的功能,例如基于网络的基因集功能注释、多重 CCA(典型相关分析)多组学数据聚类,以及基于集成方法的多组学分类,这具有更加独特的优势。
本文将仅对该工具的主要部分,即因果 WGCNA 分析进行示例演示。如果铁子们感兴趣的话,可以自行翻阅相关文档。
github 链接:
https://github.com/yuabrahamliu/CWGCNA
0. R包下载
用户可以通过以下代码安装或者引用,需要注意的是,CWGNCA 要求 R 语言版本大于等于 4.2.3:
# 从GitHub上安装该包
library(devtools)
install_github("yuabrahamliu/CWGCNA")
1. 数据导入与处理
本教程将使用随 CWGCNA 包一起提供的内置数据。数据包含了来自359个人类胎盘样本的DNA甲基化(DNAm)数据特征矩阵,这些样本基于Illumina 27K和450K平台,收集自10个不同的GEO数据集(当然,你也可以根据自己的研究类型输入其它类型的数据,处理方案有所差异)。
library(CWGCNA)
# 读入特征矩阵,这里是beta(你也可以使用其它类型的数据)
betas <- system.file("extdata", "placentabetas.rds", package = "CWGCNA")
betas <- readRDS(betas)
# 读入性状数据
pds <- system.file("extdata", "placentapds.rds", package = "CWGCNA")
pds <- readRDS(pds)
betas特征矩阵的列表示样本,行表示DNAm探针。矩阵共包含 18626 个探针,它们是各样本间共享的高质量探针。探针值为beta值,即甲基化的程度(如果你是基因表达矩阵,则为基因的表达量),已进行批次效应校正。在 359 个样本中,258 个是正常样本,剩下的101个是由胎盘异常引起的先兆子痫孕期并发症患者样本。
除了betas矩阵以外,同时需要提供的数据框还有 359 个样本的性状数据pds。性状数据包括:
-
样本ID:sampleid列,对应beta值矩阵的列名;
-
样本的分组:Group列,对应样本的先兆子痫/对照状态;
-
样本的妊娠周数:Gestwk列;
-
婴儿性别:Babygender列;
-
母亲种族:Ethnicity列;
-
样本的原始GEO数据集ID:Dataset列,对应样本在 GEO数据库的原始ID。
简单看一下示例数据:
#betas特征矩阵,The DNAm betas matrix
betas[1:6,1:6]
#> GSM788417 GSM788419 GSM788420 GSM788421 GSM788414 GSM788415
#> cg00000292 0.65961366 0.67591141 0.65709651 0.66077820 0.66847653 0.67436406
#> cg00002426 0.53516824 0.53883284 0.53683120 0.53990206 0.53804637 0.53543344
#> cg00003994 0.17674229 0.16432771 0.16631494 0.16803355 0.16416337 0.16852233
#> cg00007981 0.03553198 0.02934814 0.03189098 0.02765783 0.02768899 0.02764183
#> cg00008493 0.43619912 0.42998023 0.43761396 0.44973257 0.46281094 0.45935308
#> cg00008713 0.07431056 0.06367040 0.06764089 0.06218142 0.05267028 0.05397539
#性状数据,The metadata for the samples
head(pds)
#> sampleid Group Gestwk Babygender Ethnicity Dataset
#> 1 GSM788417 Control 8 M White GSE31781
#> 2 GSM788419 Control 8 M White GSE31781
#> 3 GSM788420 Control 8 M White GSE31781
#> 4 GSM788421 Control 9 M White GSE31781
#> 5 GSM788414 Control 12 F Asian GSE31781
#> 6 GSM788415 Control 12 M White GSE31781
# 样本分组
table(pds$Group)
#>
#> Control Preeclampsia
#> 258 101
# 样本来源的gse编号
table(pds$Dataset)
#>
#> GSE100197 GSE125605 GSE31781 GSE36829 GSE59274 GSE69502 GSE73375 GSE74738
#> 65 41 30 48 23 16 36 28
#> GSE75196 GSE98224
#> 24 48
2. 混杂因素的分析
首先使用包中的featuresampling函数,检查betas矩阵中的甲基化 beta 值与pds性状数据中的表型变量之间的关系。这个步骤执行 III 型方差分析(ANOVA),用于测量pds性状数据中各种表型变量(如分组、年龄、性别等)对特征 beta 值数据集所解释的方差。具体的执行命令与参数注释如下:
# 下方命令所需时间小于一分钟
top10k <- featuresampling(betas = betas, # 特征矩阵
topfeatures = 10000, # 仅选择前10000个最具变异性的DNAm探针构成的数据矩阵
variancetype = "sd", # 定义如何计算方差
threads = 6) # 并行化的线程数量
储存结果的对象top10k是一个列表(展示如下)。其$betas部分包含仅由前10000个最具变异性的DNAm探针构成的数据矩阵,另一个$varicanceres部分则记录了由featuresampling计算的原始 18626 个探针的标准偏差。
top10k$betas[1:6,1:6]
#> GSM788417 GSM788419 GSM788420 GSM788421 GSM788414 GSM788415
#> cg00000292 0.6596137 0.6759114 0.6570965 0.6607782 0.6684765 0.6743641
#> cg00002426 0.5351682 0.5388328 0.5368312 0.5399021 0.5380464 0.5354334
#> cg00003994 0.1767423 0.1643277 0.1663149 0.1680336 0.1641634 0.1685223
#> cg00008493 0.4361991 0.4299802 0.4376140 0.4497326 0.4628109 0.4593531
#> cg00013618 0.7859429 0.7868591 0.7933922 0.7958716 0.8006630 0.8118193
#> cg00014837 0.8522323 0.8525327 0.8516924 0.8546466 0.8610099 0.8546916
dim(top10k$betas)
#> [1] 10000 359
head(top10k$varicanceres)
#> SD
#> cg03729431 0.1848922
#> cg16063666 0.1643842
#> cg05790038 0.1456048
#> cg03316864 0.1454261
#> cg11009736 0.1393115
#> cg20322876 0.1348288
接下来,使用这个函数的$betas进行ANOVA分析:
#This command will take < 1 min
anovares <- featuresampling(betas = top10k$betas, # 使用 betas
pddat = pds, # 性状数据
anova = TRUE, # 执行 ANOVA
plotannovares = TRUE, # 绘制数据的ANOVA结果
featuretype = "probe",
plottitlesuffix = "placenta", # 显示在图表标题中的字符
titlesize = 18,
textsize = 16,
threads = 6)
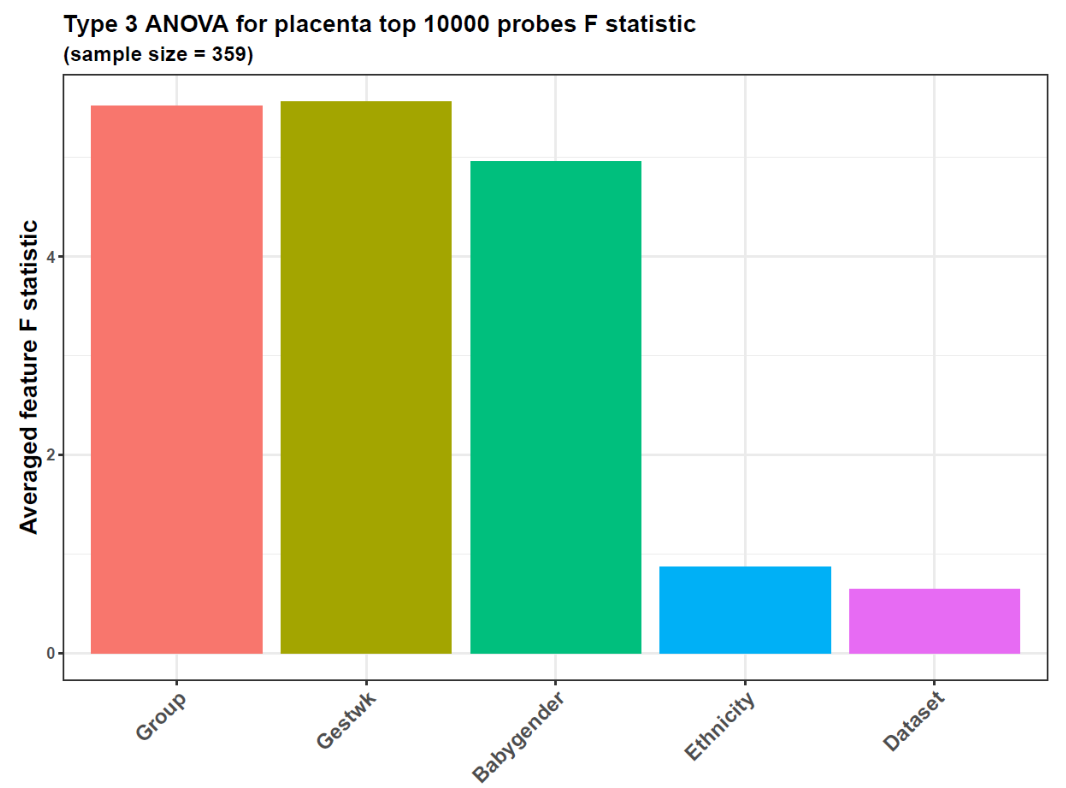
结果如上图,featuresampling 返回数据中的每个探针都将通过ANOVA,其结果F统计量、MSS(均方和)和p值将包含在列表anovares的$varianceres部分。
从图表中可以看出,先兆子痫/对照样本组、妊娠周数和婴儿性别解释了最多的方差,这意味着该数据集适合检查先兆子痫/对照样本组之间的 DNAm 差异。然而,妊娠周数和婴儿性别的方差也很高,F值>1。因此,它们是先兆子痫分析的混杂因素,需要调整它们的方差。
3. 因果WGCNA分析
接下来,对数据进行因果 WGCNA 分析。然而,在此之前,使用函数probestogenes将 DNAm 探针的 beta 值统一到基因。参数 group450k850k意味着,对于特定基因,将位于其 TSS200、TSS1500 和第一个外显子区域的探针选出,并取它们的平均值作为该基因的 beta 值 。
# This command will take < 1 min
betasgene <- probestogenes(betadat = betas, group450k850k = c("TSS200", "TSS1500", "1stExon"))
然后,使用另一个函数diffwgcna进行因果WGCNA分析。下面详细介绍一下这个函数的参数:
参数dat接受 beta 值矩阵,pddat 接受 pds 数据框。参数responsevarname和confoundings很重要,因为前者可以指定pds 中的哪一列是目标变量,后者可以指定需要调整的混杂因素。Group传递给前者,而c("Gestwk", "Babygender")传递给后者。它们都对应 pds 中的列名,是进行因果WGCNA分析所必需的。
由于已经将 DNAm 探针名称转换为基因名称,所以将参数featuretype设为“gene”。随后,还将参数topvaricancetype设为“sd”,并将topvaricance设为5000,意味着将在数据中最具变异性的前5000个基因上进行分析(标准偏差最高的那些)。
函数diffwgcna在传统的WGCNA框架内进行因果推断。因此,有必要调用传统的 WGCNA 模块。将向量seq(1, 20, 1)传递给powers参数,这样可以通过网格搜索从1到20选择WGCNA的最佳软阈值。另一个参数rsqcutline设为0.8,目标最小无标度拓扑拟合指数是0.8。
参数mediation很重要,因为它决定在WGCNA流程中是否需要执行因果推断的核心——中介分析。对于每个WGCNA模块,它会构建中介模型以测试两个因果方向。一个方向是“模块→模块基因→先兆子痫”,另一个方向是“先兆子痫→模块基因→模块”。因此,可以确定疾病使得模块变化,或模块驱动疾病。
将参数topn设为1,因此因果推断仅针对最具差异性的1个WGCNA模块及其特征执行。默认情况下,此参数为 NULL,在这种情况下,所有差异模块都会进行因果推断,但分析所有模块会耗费时间。
如果参数plot设置为TRUE,将生成若干图表来展示结果,并且传递给titleprefix参数的字符会显示在图表标题中。参数labelnum表示在 limma 回归和因果推断结果中标记在图表中的前几个特征。可以将其设置为 NULL,使没有特征名称被标记。在这里将其设置为5。参数annotextsize可控制这些特征标签的字体大小。
参数pvalcolname设置为“adj.P.Val”,pvalcutoff设为0.05,表示limma 回归识别的差异模块和基因应该有调整后的p值<0.05。由于这里的数据值是 DNAm beta值,所以将参数isbetaval设为TRUE。参数diffcutoff定义模块特征基因差异的阈值,以选择差异模块,而参数absxcutoff定义模块内基因的 log2FC 阈值,以选择差异基因。由于基因值是beta值,limma 的 log2FC 结果表示组间的 beta 值差异。
这个 WGCNA 分析耗时较长(约25分钟):
#This command will take ~ 25 min
cwgcnares <- diffwgcna(dat = betasgene,
pddat = pds,
responsevarname = "Group",
confoundings = c("Gestwk", "Babygender"),
featuretype = "gene",
topvaricancetype = "sd",
topvaricance = 5000,
powers = seq(1, 20, 1),
rsqcutline = 0.8,
mediation = TRUE,
topn = 1,
plot = TRUE,
titleprefix = "Placenta",
labelnum = 5,
titlesize = 17,
textsize = 16,
annotextsize = 5,
pvalcolname = "adj.P.Val",
pvalcutoff = 0.05,
isbetaval = TRUE,
absxcutoff = 0,
diffcutoff = 0,
threads = 6)
结果cwgcnares是一个包含多个部分的列表,其中名为$limmares的部分是对所有WGCNA模块的 limma 结果,函数生成的柱状图(如下展示)便是基于此结果。它显示数据包含从 ME1 到 ME11 的11个模块,其中7个模块在先兆子痫/对照组之间具有显著差异的特征基因。
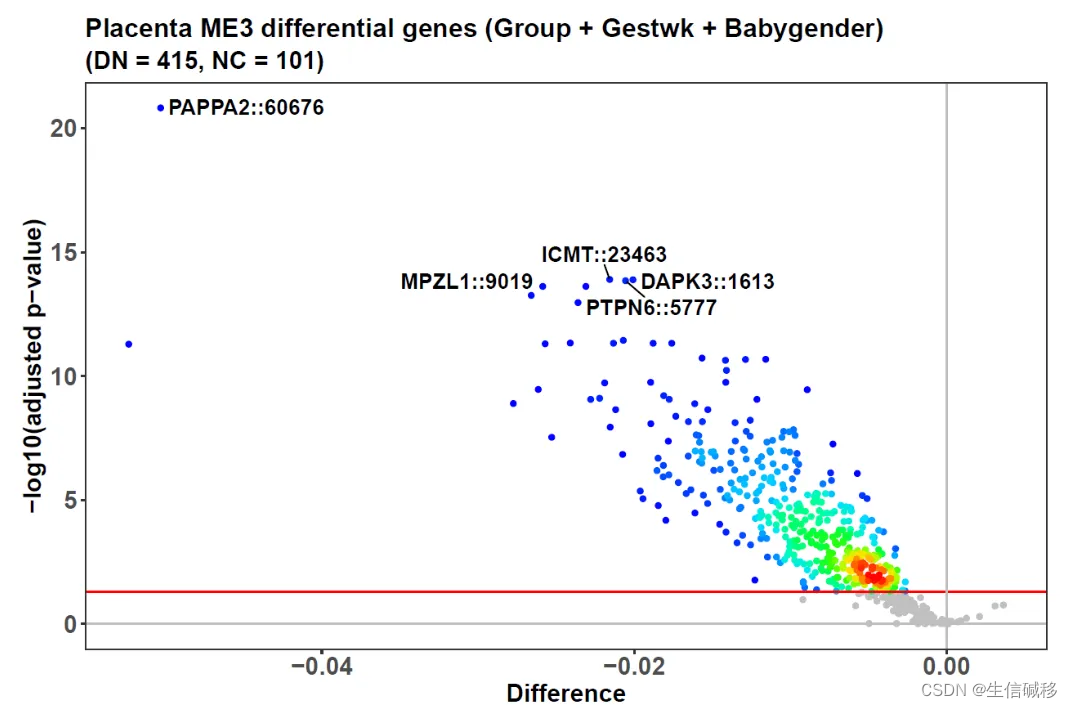
此外,diffwgcna还调用 limma 选择每个模块内的差异特征。例如下面这张ME3的火山图显示,与对照组相比,先兆子痫患者组中 516 个基因中的 415 个甲基化降低。$melimmares部分包含每个显著差异模块内的结果。
由于参数mediation设为TRUE,且参数topn设为1,最具差异性的模块ME3的因果推断结果已返回,$mediationres 部分记录了这个结果。相应地,ME3 获得了额外的火山图来显示其中介结果(如下图展示)。在这第二个火山图中,319 个蓝点所指的基因在“先兆子痫→ME3基因→ME3”因果方向(反向)起中介作用,而只有 2 个红点所指的基因在“ME3→ME3基因→先兆子痫”方向(正向)起中介作用。y轴和x轴分别显示筛选差异基因时的-log10(调整后的p值)和先兆子痫/对照beta值差异。
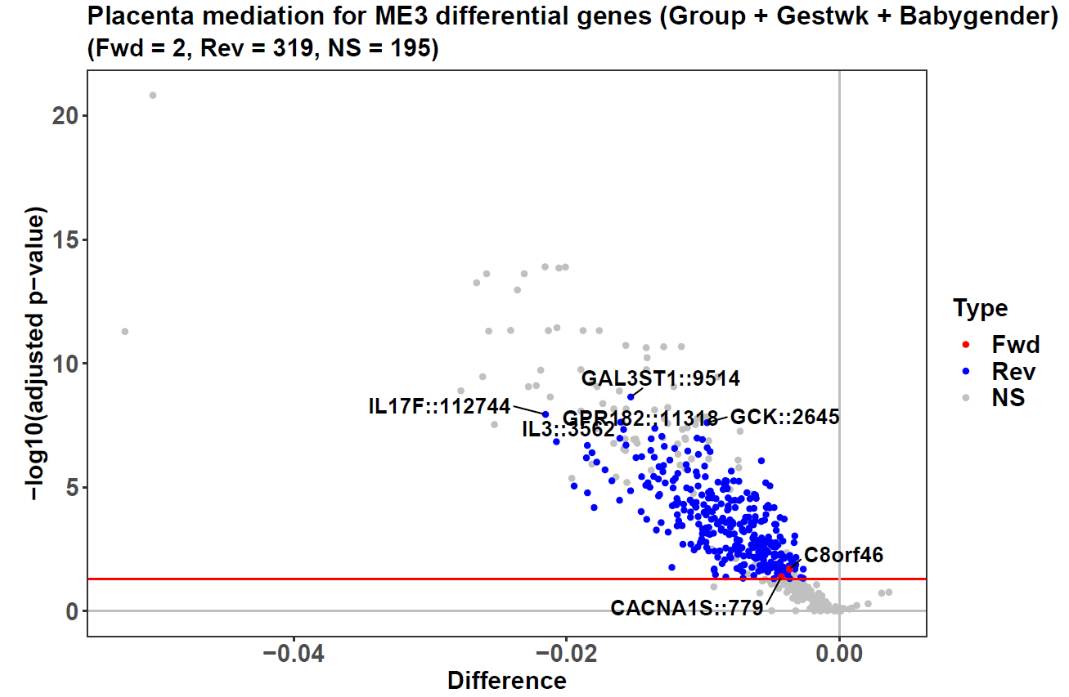
这 319 个基因与先兆子痫中的炎症应激有关,例如 ME3 中发现的 top 基因 IL3 和 IL17F,它们都是炎症相关的细胞因子。由于多数基因的因果方向是从先兆子痫到ME3,因此可以得出结论,炎症是这种疾病的结果而非原因。
同时,CACNA1S 是从 ME3 到先兆子痫的因果方向中的 top 基因。该基因在胎盘中表达,编码负责血管收缩的钙通道。鉴于先兆子痫是一种妊娠期高血压疾病,CACNA1S 对其的驱动作用非常明显。此外,该基因是治疗先兆子痫的小分子药物苯磺酸氨氯地平的靶标,这进一步证明了这里的因果推断是合理的。